快速入门
本章将介绍ARES的各种功能的基本使用,具体包括: scf自洽计算、结构优化计算、能带计算、态密度计算、杂化泛函计算、分子动力学模拟、晶格动力学计算。ARES软件的参数大致可以分为:与结构相关,与计算性质相关,与计算精度相关,与收敛相关,并且大部分都有默认值。本章筛选部分参数进行介绍,参数列表及详情另见参数说明部分。
scf自洽计算
自洽计算能得到特定晶体的电荷密度和波函数文件,电荷密度文件用于后续计算该体系的能带、态密度等电子结构性质。特别需要注意是:自洽与能带、态密度等电子结构性质计算是有先后顺序的,必须先进行自洽计算得到电荷密度才能进一步计算能带、态密度等电子结构性质。
示例输入(ares.in)
INPUT_PARAMETERS
cal_type = scf
sym = 2
ecutwfc = 820
basis = pw
diago = dav
smearing = mp
sigma = 0.01
mixing_type = broyden
mixing_beta = 0.7
scf_energy_thr = 1e-07
force_cal = 1
stress_cal = 1
out_chg = 1
init_chg = atomic
pseudo_dir = path/to/your/PSP
pseudo_file = Rb.upf B.upf C.upf
kpar = 2
要点说明
cal_type: 计算类型,本例为scf;sym: 设置对称性,0:不考虑对称性,1:仅考虑时间反演对称性,2:考虑所有的对称性;ecutwfc:平面波截断能,注意单位为eV;basis: 基组类型,pw为平面波基组;diago: 迭代求解算法,可选cg或dav;smearing: 电子态展宽方法;sigma: 电子态展宽参数;mixing_type: 自洽迭代电荷密度混合方法;mixing_beta: 当前步电子密度贡献;scf_energy_thr: 能量收敛阈值(eV);force_cal: 是否计算受力;stress_cal: 是否计算应力;out_chg: 是否输出电荷密度;init_chg: 电荷密度初始化方法, file为从文件读取;pseudo_dir: 赝势文件路径,请将"path/to/your/PSP"修改为赝势路径;pseudo_file: 元素赝势文件名,注意与ares.cell中元素名顺序一致;kpar: k点并行参数;
结构文件示例(ares.cell)
Rb B C # 提示
1.00000000 # 晶格缩放比例
#晶格矢量
4.9319105017 0.0000000000 -0.0000000000
0.0000000000 4.9319105017 -0.0000000000
-0.0000000000 -0.0000000000 4.9319105017
Rb B C #原子类型,注意与输入文件赝势顺序对应
2 6 6 #原子数量
Direct #坐标类型(Direct/Cartesian)
#原子坐标
0.0000000000 0.0000000000 0.0000000000
0.5000000000 0.5000000000 0.5000000000
0.2500017006 0.5000000000 0.0000000000
0.7499982844 0.5000000000 0.0000000000
0.0000000000 0.2500017006 0.5000000000
0.5000000000 0.7499982844 0.5000000000
0.5000000000 0.0000000000 0.2500017006
0.5000000000 0.0000000000 0.7499982844
0.2499977945 0.0000000000 0.5000000000
0.7500217550 0.0000000000 0.5000000000
0.5000000000 0.2499977945 0.0000000000
0.5000000000 0.7500217550 0.0000000000
0.0000000000 0.5000000000 0.5000000000
0.5000000000 0.5000000000 0.0000000000
K点文件示例(ares.kpoints)
K_POINTS
0
Gamma
8 8 8 0 0 0
K_POINTS: K点设置的标识符,表明这是一个K点相关的配置文件0: 表示使用自动生成的 K 点网格模式。在 ARES 中,这个参数指定 K 点的生成方式,0 代表使用自动生成模式。Gamma: 表示以 Gamma 点为中心生成 K 点网格。8 8 8 0 0 0:K点网格设置前三个数字(8 8 8)定义了沿三个倒格子方向的 K 点网格数量,即每个方向上只取 8 个K 点
后三个数字(0 0 0)表示 K 点网格的偏移量,这里为 0 表示没有偏移
计算过程分析
内容见于输出文件 ares.log:
#### Start SCF
| ITER | ETOT/eV | EDIFF/eV | DRHO | TIME/s |
|--------|-----------------|-----------------|-----------------|-----------------|
| 1 | -2.660385e+03 | 0.000000e+00 | 3.430202e+00 | 7.887309 |
| 2 | -2.665094e+03 | -4.709416e+00 | 3.250900e+00 | 2.776408 |
| 3 | -2.674068e+03 | -8.973706e+00 | 1.153292e+00 | 2.368413 |
| 4 | -2.675817e+03 | -1.749436e+00 | 2.443068e-02 | 1.945961 |
| 5 | -2.675871e+03 | -5.403346e-02 | 8.827870e-03 | 3.269010 |
| 6 | -2.675885e+03 | -1.394592e-02 | 6.387593e-03 | 2.601838 |
| 7 | -2.675898e+03 | -1.262792e-02 | 3.836782e-04 | 2.142792 |
| 8 | -2.675899e+03 | -1.221807e-03 | 4.203134e-05 | 2.448310 |
| 9 | -2.675899e+03 | -8.774950e-05 | 6.382143e-06 | 2.479172 |
| 10 | -2.675899e+03 | -1.280452e-05 | 6.678881e-07 | 2.608026 |
| 11 | -2.675899e+03 | -1.937245e-06 | 1.460633e-07 | 2.673662 |
| 12 | -2.675899e+03 | -3.547363e-07 | 1.236370e-08 | 2.077668 |
| 13 | -2.675899e+03 | -2.982982e-08 | 8.932649e-10 | 2.929819 |
TOTAL-PRESSURE: 85.321298 GPa
ITER: 电子步数ETOT/eV: 体系总能EDIFF/eV: 能量差DRHO: 电荷密度差TIME/s: 单电子步计算时间TOTAL-PRESSURE: 体系压强
更加详细的电子步内容见于 ares_output.dat ,以其中一步为例:
#### PW
|-----------|-------|
| ION | 1 |
| ELEC | 12 |
SCF Energy
|-------------|--------------|
| KS | -2675.899161 |
| KS(sigma:0) | -2675.900513 |
| harris | -2675.896520 |
| hartree | 436.239420 |
| xc | -686.581381 |
| ewald | -2521.397877 |
| entropy | 0.004054 |
| fermi | 11.372933 |
能量单位eV
ION: 离子步ELEC: 电子步KS: 本电子步 Kohn–Sham 总能量KS(sigma→0): 本电子步通过展宽修正得到的零温近似能量。harris: Harris 估计能量hartree: 电子间库仑能xc: 交换相关能ewald: 离子间 Ewald 能entropy: 电子熵fermi: 费米能级
计算结果分析
内容见于输出文件 ares_output.dat:
体系能量
### Final Total Energy (eV)
|------------------------|--------------|
| energy toten | -2675.899161 |
| energy without entropy | -2675.903214 |
| energy(sigma->0) | -2675.900512 |
| enthalpy toten | -2675.899161 |
| PV | 0.000000 |
| fermi | 11.372913 |
energy toten: 体系的 Kohn–Sham 总能量energy without entropy: 从自由能中去掉熵后的能量energy(sigma→0): 通过展宽修正得到的零温近似能量。enthalpy toten: 体系焓值PV: 压强乘以体积的能量项fermi: 费米能级
体系原子受力表
#### TOTAL-FORCE
|------|-----|----------------|----------------|----------------|
| Rb | 1 | -0.000515 | -0.415828 | 0.000000 |
| Rb | 2 | 0.000472 | -67.007144 | 0.000000 |
| B | 1 | -14.206187 | 0.082065 | 0.000000 |
| B | 2 | 14.205979 | 0.082063 | 0.000000 |
| B | 3 | -0.000421 | -16.443898 | 0.000000 |
| B | 4 | 0.000399 | 70.984356 | 0.000000 |
| B | 5 | 0.000344 | 0.719383 | 0.634403 |
| B | 6 | 0.000344 | 0.719383 | -0.634403 |
| C | 1 | 4.237854 | 0.250927 | 0.000000 |
| C | 2 | -4.238465 | 0.250618 | 0.000000 |
| C | 3 | 0.000178 | -14.175339 | 0.000000 |
| C | 4 | 0.000052 | 12.927439 | 0.000000 |
| C | 5 | -0.000267 | 12.208165 | 0.000000 |
| C | 6 | 0.000232 | -0.182190 | 0.000000 |
每行对应一个原子的三维力分量,单位eV/Å,原子受力较大,可通过结构优化减小原子受力,获得稳定结构
体系应力张量
#### TOTAL-STRESS (KBAR)
|---|------------|-------------|------------|
| x | 550.373429 | 0.002079 | 0.000000 |
| y | 0.002079 | 1784.549035 | 0.000000 |
| z | 0.000000 | 0.000000 | 224.716471 |
单位为 kbar
计算时间
| Parameter | Value |
|-------------|--------------------------|
| Start Time | Wed Oct 15 13:40:31 2025 | #任务开始时间
| Finish Time | Wed Oct 15 13:41:10 2025 | #任务结束时间
| Total Time | 0 h 0 mins 39 secs | #任务总时间
结构优化(cell-relax)计算
示例输入(ares.in)
INPUT_PARAMETERS
# control
cal_type = cell-relax # 晶胞弛豫计算
# system
basis = pw # 平面波基组
dft_functional = PBE # 交换-相关泛函
sym = 2 # 考虑所有对称性操作
ecutwfc = 980 # 截断能 (eV)
smearing = gauss # 电子展宽类型
sigma = 0.01 # 展宽参数 (eV)
# electron
iscf_iter = 100 # 最大 SCF 迭代步数
diago = dav # 本征值求解算法(Davidson)
scf_energy_thr = 1e-7 # SCF 收敛阈值 (eV)
# ion
iopt_iter = 500 # 最大原子弛豫步数
relax_method = bfgs # 弛豫算法(BFGS)
pressure = 850 # 外压 (kbar)
force_thr = 0.001 # 力收敛阈值 (eV/Å)
# pseudopotential
pseudo_dir = ./
pseudo_file = Rb.upf B.upf C.upf
# parallel
kpar = 2 # K 点并行数
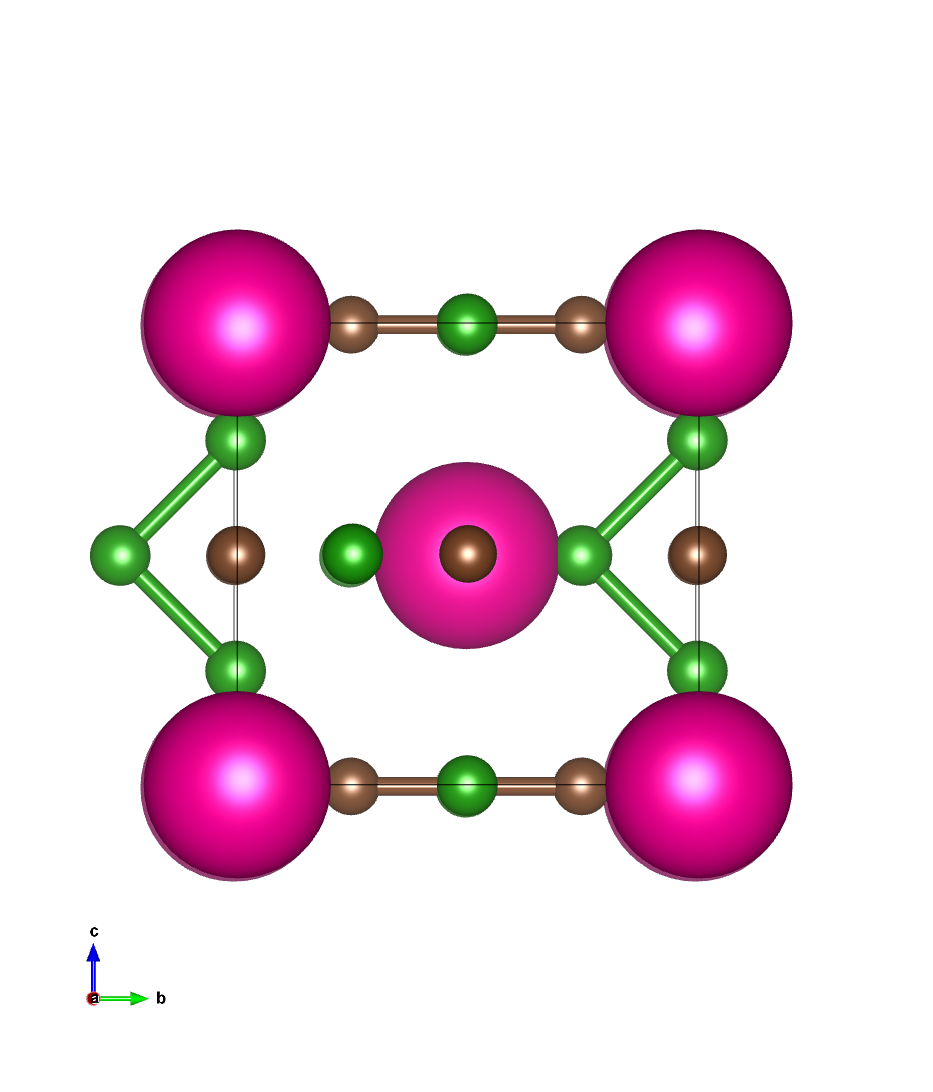
结构文件示例(ares.cell)
与scf计算结构相同
结构图像:
K点文件示例(ares.kpoints)
与scf计算结构相同
计算结果分析
优化后结构输出为``opt.cell`` :
Rb B C
1.0000000
4.1053374808 0.0004776580 0.0000000000
-0.0005005063 5.7159472401 0.0000000000
0.0000000000 0.0000000000 4.3918395751
Rb B C
2 6 6
Direct
0.9999911408 0.8504753082 0.0000000000
0.4999824298 0.4578075248 0.5000000000
0.2039656396 0.4218354069 0.0000000000
0.7960843244 0.4218432819 0.0000000000
0.9999929598 0.2148762744 0.5000000000
0.5000060276 0.8524780932 0.5000000000
0.4999981578 0.0489430411 0.2257809801
0.4999981578 0.0489430411 0.7742190049
0.2290578932 0.0188894548 0.5000000000
0.7709305818 0.0188967355 0.5000000000
0.5000379889 0.2416338360 0.0000000000
0.4999877373 0.8344268059 0.0000000000
0.9999812141 0.4726895084 0.5000000000
0.5000052816 0.5962812222 0.0000000000
原子受力:
#### TOTAL-FORCE
|------|-----|----------------|----------------|----------------|
| Rb | 1 | 0.000317 | -0.003554 | 0.000000 |
| Rb | 2 | -0.000145 | 0.001160 | 0.000000 |
| B | 1 | 0.003040 | 0.000604 | 0.000000 |
| B | 2 | -0.003427 | -0.000202 | 0.000000 |
| B | 3 | -0.000105 | 0.001982 | 0.000000 |
| B | 4 | 0.000134 | 0.006508 | 0.000000 |
| B | 5 | -0.000109 | -0.004278 | -0.003269 |
| B | 6 | -0.000109 | -0.004278 | 0.003269 |
| C | 1 | 0.004586 | -0.000236 | 0.000000 |
| C | 2 | -0.004109 | -0.001005 | 0.000000 |
| C | 3 | -0.000694 | 0.005756 | 0.000000 |
| C | 4 | 0.000394 | 0.002474 | 0.000000 |
| C | 5 | -0.000156 | -0.004424 | 0.000000 |
| C | 6 | 0.000382 | -0.000507 | 0.000000 |
原子受力均小于0.01eV/Å,优化效果明显。
体系应力张量:
#### TOTAL-STRESS (KBAR)
|---|------------|------------|------------|
| x | 850.220252 | 0.000397 | 0.000000 |
| y | 0.000397 | 850.102855 | 0.000000 |
| z | 0.000000 | 0.000000 | 849.905861 |
TOTAL-PRESSURE: 85.007632 GPa
结构图像:
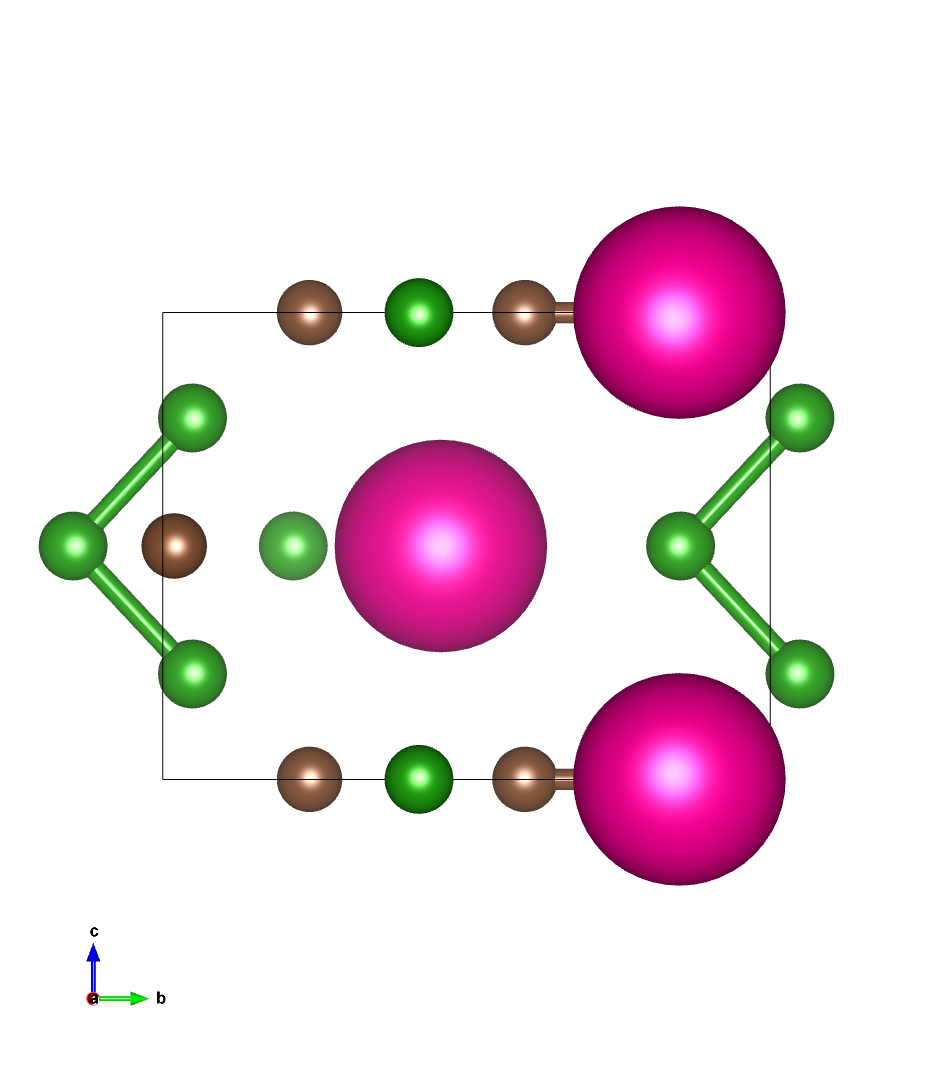
QE优化结果对比:
结构信息:
CELL_PARAMETERS (angstrom)
4.113885685 0.000057134 0.000000000
-0.000305051 5.733964505 0.000000000
0.000000000 0.000000000 4.371866607
ATOMIC_POSITIONS (crystal)
Rb 0.0000020507 -0.1509811544 0.0000000000
Rb 0.5000895283 0.4575416759 0.5000000000
B 0.2037869098 0.4220371979 0.0000000000
B 0.7961192170 0.4220257130 0.0000000000
B 0.0000491185 0.2144516077 0.5000000000
B 0.5000167462 0.8512787771 0.5000000000
B 0.4999752355 0.0495590891 0.2251955623
B 0.4999752355 0.0495590891 0.7748044227
C 0.2300709641 0.0189337926 0.5000000000
C 0.7699131361 0.0189861225 0.5000000000
C 0.4999328266 0.2427968845 0.0000000000
C 0.5000132878 0.8340810893 0.0000000000
C 0.0000960177 0.4729054562 0.5000000000
C 0.4999792607 0.5968441939 0.0000000000
End final coordinates
原子受力:
Forces acting on atoms (cartesian axes, Ry/au):
atom 1 type 1 force = -0.00002220 0.00007026 0.00000000
atom 2 type 1 force = -0.00000516 -0.00020711 0.00000000
atom 3 type 2 force = 0.00011311 0.00001780 0.00000000
atom 4 type 2 force = -0.00011244 0.00003815 0.00000000
atom 5 type 2 force = 0.00000141 -0.00034248 0.00000000
atom 6 type 2 force = -0.00001319 0.00002504 0.00000000
atom 7 type 2 force = 0.00002918 0.00008001 -0.00014503
atom 8 type 2 force = 0.00002918 0.00008001 0.00014503
atom 9 type 3 force = -0.00001840 0.00005946 0.00000000
atom 10 type 3 force = 0.00004094 0.00001843 0.00000000
atom 11 type 3 force = 0.00002831 -0.00027660 0.00000000
atom 12 type 3 force = -0.00004423 0.00119145 0.00000000
atom 13 type 3 force = -0.00000159 0.00027555 0.00000000
atom 14 type 3 force = -0.00002491 -0.00103000 0.00000000
Total force = 0.001701 Total SCF correction = 0.000037
体系应力张量:
Computing stress (Cartesian axis) and pressure
total stress (Ry/bohr**3) (kbar) P= 853.22
0.00577472 -0.00000013 0.00000000 849.49 -0.02 0.00
-0.00000013 0.00585043 0.00000000 -0.02 860.63 0.00
0.00000000 0.00000000 0.00577506 0.00 0.00 849.54
优化结构图像:
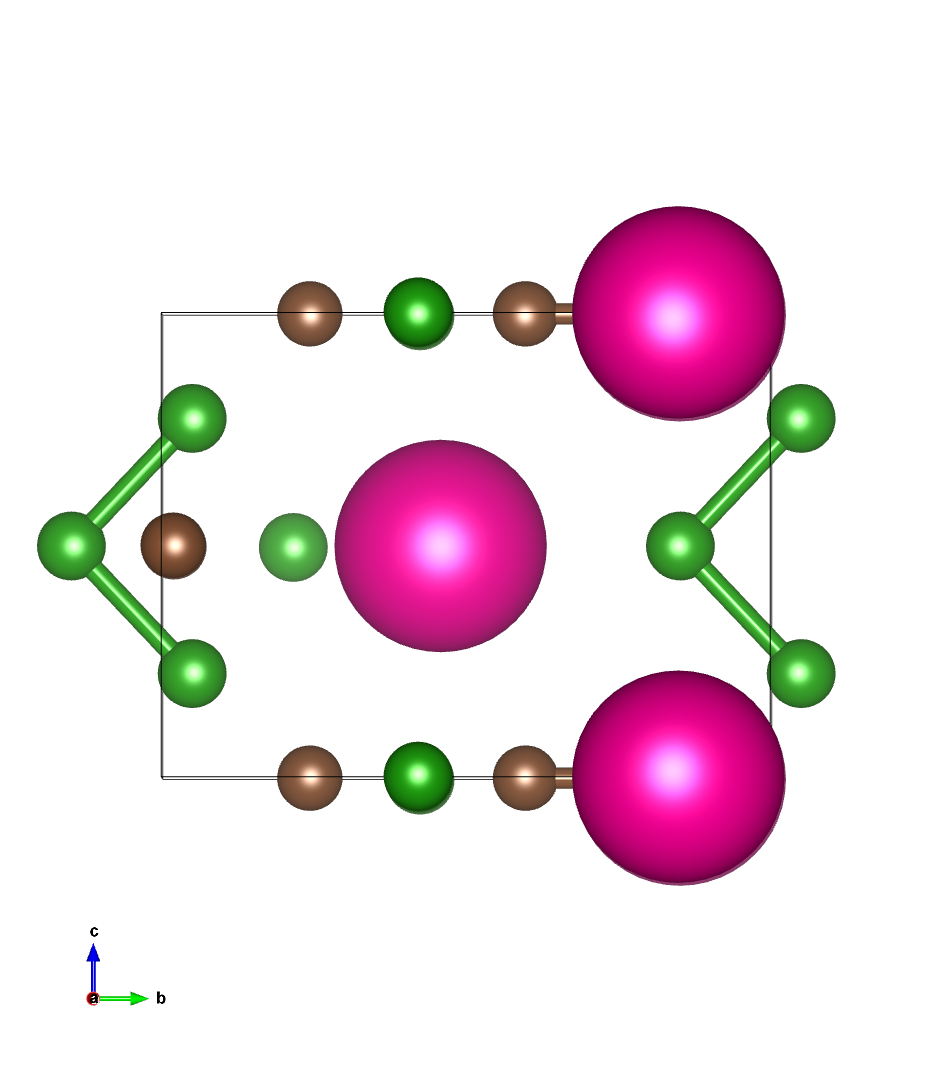
通过比较可见,ARES与QE优化后的结构与原子受力等精度一致
结构弛豫计算示例2:单质硅
本节将以单质硅为例子,介绍ARES中的结构弛豫计算。
结构弛豫计算输入文件
输入文件包含参数文件 ares.in,结构文件 ares.cell,k点文件 ares.kpoints 和赝势文件(模守恒赝势,格式为.UPF)。
ares.in 文件内容如下:
INPUT_PARAMETERS
# control
cal_type = cell-relax
# system
basis = pw
dft_functional = PBE
sym = 2
ecutwfc = 980 #eV
smearing = gauss
sigma = 0.01
# electron
iscf_iter = 100
diago = dav
scf_energy_thr = 1e-7
# ion
iopt_iter = 100
relax_method = bfgs
pressure = 0.0 #kbar
force_thr = 0.001 #eV/Å
# pseudopotential
pseudo_dir = ./
pseudo_file = Si.upf
# parallel
kpar = 4
ares.in 中的参数大致可以分为六个部分
第一部分指定计算类型,以及输出控制(relax计算输出控制为默认不额外设置)
cal_type:设置计算类型,这里选择晶格可变的结构弛豫计算。
第二部分指定系统相关控制参数,涉及基组、泛函、对称性等
basis:设置基组,这里选择平面波(倒空间)基组。
dft_functional:设置泛函,这里选择PBE泛函。
sym:设置对称性,这里选择2,即应用全部对称性。
ecutwfc:设置波函数动能截断,这里选择980 eV。
smearing:设置在布里渊区积分中,处理能带占据数的展宽函数,这里选择高斯展宽。
sigma:设置高斯平滑宽度,这里选择0.01 Ry。
第三部分指定电子自洽迭代相关控制参数,迭代步数、对角化方法、收敛参数等
iscf_iter:设置自洽迭代步数,这里选择100步。
diago:设置对角化方法,这里选择Davidson方法。
scf_energy_thr:设置自洽能量收敛阈值,这里选择1e-7 eV。
第四部分指定离子弛豫迭代相关控制参数,迭代步数、优化方法、收敛参数等
iopt_iter:设置离子弛豫迭代步数,这里选择100步。
relax_method:设置优化方法,这里选择BFGS方法。
pressure:设置压力,这里选择0.0kbar,即常压。
force_thr:设置力收敛阈值,这里选择0.001eV/Å。
第五部分指定赝势文件信息
pseudo_dir:设置赝势文件所在目录,这里选择当前目录。
pseudo_file:设置赝势文件名,这里选择Si.upf。
第六部分指定并行控制参数
kpar:设置k点并行数,这里选择4。
ares.cell 文件参考如下:
Si
1.0
0.0000000000 2.7153607857 2.7153607857
2.7153607857 0.0000000000 2.7153607857
2.7153607857 2.7153607857 0.0000000000
Si
2
Direct
0.0100000000 0.0000000000 0.0000000000
0.2500000000 0.2500000000 0.2500000000
结构文件格式固定:第一行为提示/命名行。第二行为晶格缩放比例,之后三行为晶格基矢矩阵,由三个基矢横向量组成,单位为Å。第六、七行为元素符号和原子数。第八行为原子坐标类型(Direct/Cartesian),此处为相对坐标;第九行开始为具体原子坐标,由三个坐标分量组成。
为体现本案例的结构弛豫功能,我们手动修改 ares.cell 文件中的原子坐标,将第一个Si原子的坐标从 (0.00, 0.00, 0.00) 改为 (0.01, 0.00, 0.00),此时结构对称性由Fd-3m下降到C2/m。
ares.kpoints 文件参考如下:
K_POINTS
0
Gamma
16 16 16 0 0 0
K 点文件格式固定:第一行为 K 点设置的标识符 K_POINTS。第二行 0 表示使用自动生成的 K 点网格模式(Monkhorst-Pack 网格),第三行 Gamma 表示移动网格中心到 Gamma 点。第四行开始为 K 点网格参数,前三个数字定义了沿三个倒格子方向的 K 点网格数,后三个数字表示 K 点网格的偏移量。
计算结果分析
根据上述的输入文件,计算完成之后将会得到 ares.log 、ares._output.dat 、opt.cell 三个文件。
ares.log 为计算日志,包含了电子自洽迭代次数、离子弛豫迭代次数、电子总能量、计算总时间等信息。
ares._output.dat 为计算的输出文件,包括计算任务信息、赝势、输入参数、初始化信息、具体的电子自洽迭代、离子弛豫迭代信息、原子受力、晶格应力、能量等。
opt.cell 为结构优化结果文件,包含了优化后的结构信息。
本案例中得到的 opt.cell 文件内容如下:
Si
1.0
0.0114851099 2.7316872491 2.7316872491
2.7378165385 0.0056029937 2.7375710576
2.7378165385 2.7375710576 0.0056029937
Si
2
Direct
0.0056698942 0.9999715149 0.9999715149
0.2543301058 0.2500284851 0.2500284851
以 1e-3 Å 误差容忍度寻找对称性,结构恢复到 Fd-3m 空间群,使用对称性规范后结构信息如下:
Si
1.0
0.0000000000 2.7347651052 2.7347651052
2.7347651052 0.0000000000 2.7347651052
2.7347651052 2.7347651052 0.0000000000
Si
2
Direct
0.0000000000 0.0000000000 0.0000000000
0.2500000000 0.2500000000 0.2500000000
在本晶格可变的结构弛豫计算案例中,可以看到人为挪动的第一个Si原子重新回到正确坐标。同时晶格参数也进行了优化,释放了晶格应力。
能带(bands)计算
运行完 scf 后,可以进行能带计算。能带计算需要指定高对称点路径,ARES 通过k点文件指定高对称点路径
示例输入(ares.in)
INPUT_PARAMETERS
cal_type = nscf
iopt_iter = 1
sym = 1
ecutwfc = 820
basis = pw
diago = dav
smearing = mp
sigma = 0.01
mixing_type = broyden
mixing_beta = 0.7
scf_energy_thr = 1e-07
cal_band = 1
nbands = 48
init_chg = file
pseudo_dir = ./
pseudo_file = Rb.upf B.upf C.upf
kpar = 2
与scf计算输入文件的不同之处:
cal_type: 计算类型设置为nscf,进行单电子步非自洽计算sym: 设为1,只考虑空间反演对称性cal_band: 设为1,输出能带本征值nbands: 能带数目init_chg: 初始电荷密度从scf计算结果读取
结构文件示例(ares.cell)
与scf计算结构相同
K点文件示例(ares.kpoints)
K_POINTS
6
Line
0.0000000000 0.0000000000 0.0000000000 40 # GAMMA
0.0000000000 0.5000000000 0.0000000000 40 # X
0.5000000000 0.5000000000 0.0000000000 40 # M
0.0000000000 0.0000000000 0.0000000000 40 # GAMMA
0.5000000000 0.5000000000 0.5000000000 40 # R
0.0000000000 0.5000000000 0.0000000000 1 # X
K_POINTS: K点设置的标识符,表明这是一个K点相关的配置文件6: 表示高对称路径K点数目Line: 表示生成K点路径。0.0000000000 0.0000000000 0.0000000000 40 # GAMMA ...:K点路径设置,需用户自行给出前三列表示高对称k点坐标
第四列表示到下一个k点的步长,本计算共201个k点
计算结果分析
band计算结果输出在``bands.dat``文件中,内容如下:
1 0.00000000 -19.8086 -14.7776 -10.7817 -6.4893 -5.0455 -4.5962 -4.3750 -3.8807 -2.1764 -0.7038 -0.2570 -0.0733 0.2445 0.7644 2.2841 2.6507 3.2846 3.8060 4.8045 ...
2 0.00253451 -19.8086 -14.7776 -10.7816 -6.4903 -5.0460 -4.5961 -4.3730 -3.8804 -2.1762 -0.7035 -0.2569 -0.0747 0.2442 0.7646 2.2829 2.6502 3.2838 3.8048 4.8069 ...
3 0.00506903 -19.8086 -14.7775 -10.7816 -6.4933 -5.0478 -4.5957 -4.3673 -3.8793 -2.1757 -0.7028 -0.2569 -0.0791 0.2431 0.7653 2.2793 2.6487 3.2815 3.8013 4.8141 ...
4 0.00760354 -19.8085 -14.7774 -10.7815 -6.4982 -5.0507 -4.5950 -4.3578 -3.8775 -2.1748 -0.7017 -0.2567 -0.0864 0.2413 0.7665 2.2734 2.6461 3.2775 3.7956 4.8261 ...
...
第一列:表示k点序号
第二列:表示k点在路径上的累计距离,用于画能带时的横坐标
第三列及之后:表示各能带在该 k 点的能量,单位eV。本计算nbands=48,故有48列能量值
能带图绘制如下:
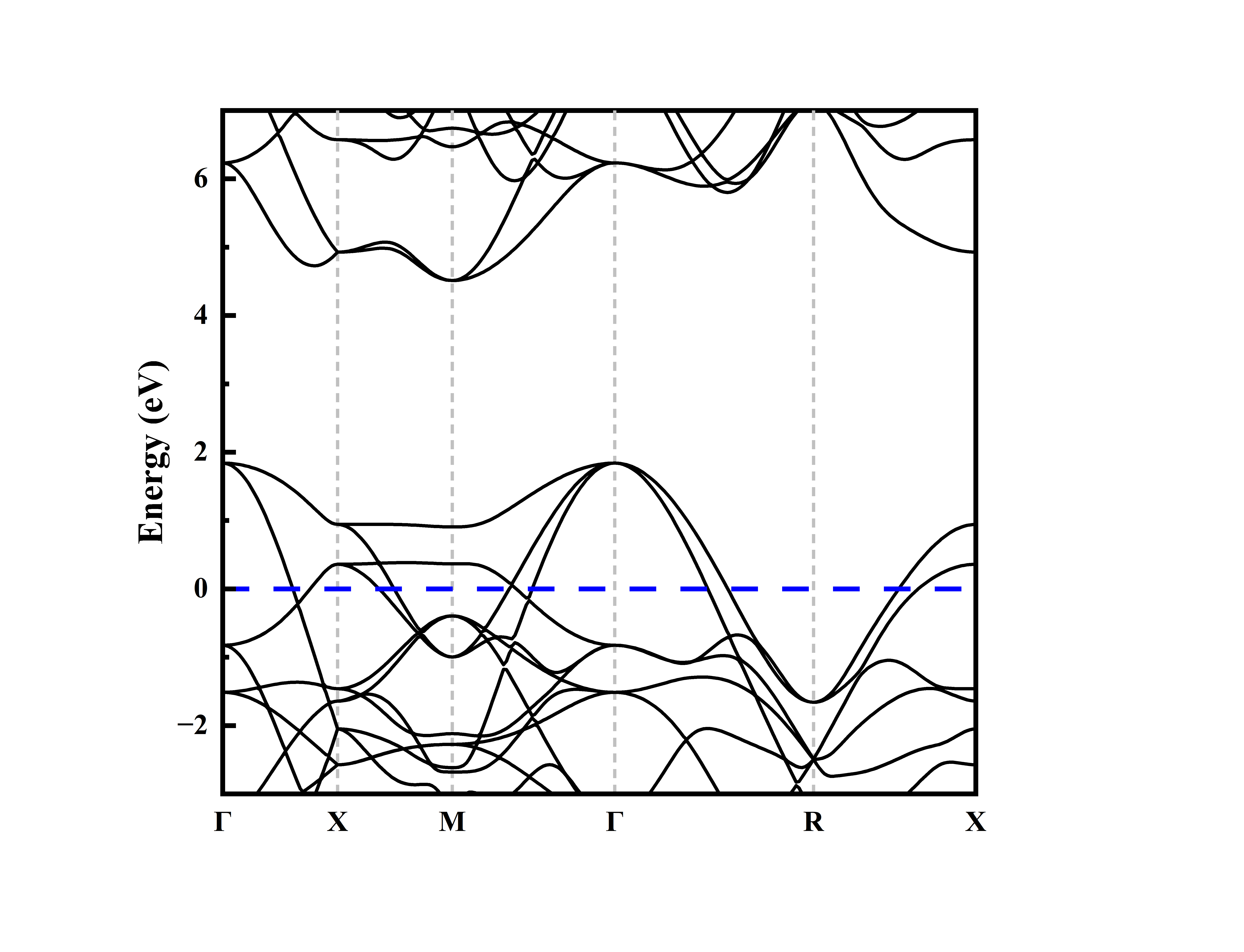
横坐标为k点路径,纵坐标为能量值
态密度(dos)计算
本节从自洽出发计算态密度
示例输入(ares.in)
INPUT_PARAMETERS
cal_type = nscf
iopt_iter = 1
sym = 2
ecutwfc = 820
basis = pw
diago = dav
smearing = mp
sigma = 0.01
mixing_type = broyden
mixing_beta = 0.7
scf_energy_thr = 1e-07
nbands = 48
init_chg = file
cal_dos = 1
pseudo_dir = ./
pseudo_file = Rb.upf B.upf C.upf
kpar = 2
dos_step = 0.01
dos_smear = 0.01
与scf,bands计算输入文件的不同之处:
cal_dos: 设置为1,开启dos计算并输出计算结果dos_step: 计算DOS时能量网格的间隔dos_smear: 对每个能级进行展宽的宽度,控制DOS曲线的光滑程度。
结构文件示例(ares.cell)
与scf计算结构相同
K点文件示例(ares.kpoints)
K_POINTS
0
Gamma
20 20 20 0 0 0
需要比scf更密的k点
计算结果分析
band计算结果输出在``dos.dat``文件中,内容如下:
### energy(eV) states/ev/u.c. elec_int
-20.004597 0.000000 0.000000
-19.994597 0.000000 0.000000
-19.984597 0.000000 0.000000
-19.974597 0.000000 0.000000
-19.964597 0.000000 0.000000
-19.954597 0.000000 0.000000
-19.944597 0.000000 0.000000
-19.934597 0.000000 0.000000
-19.924597 0.000000 0.000000
-19.914597 0.000000 0.000000
-19.904597 0.000000 0.000000
-19.894597 0.000000 0.000000
. . .
. . .
. . .
第一列为能量值,单位eV
第二列为每单位能量、每晶胞的态密度
第三列为态密度积分值(即电子累计数)
态密度图绘制如下:
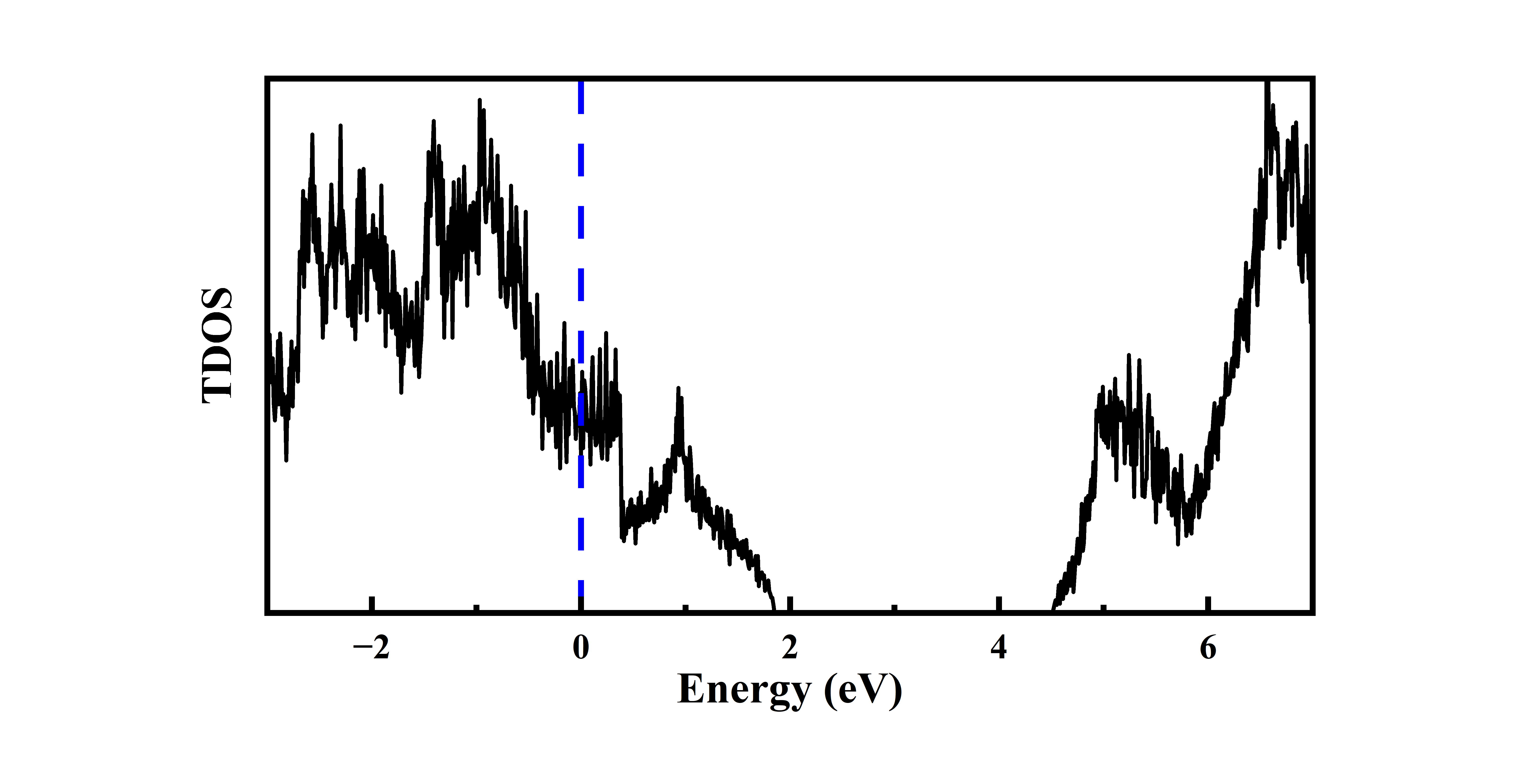
横坐标为能量值,纵坐标为态密度
杂化泛函自洽计算(HSE06)
ARES支持实空间基组的HSE06杂化泛函计算,本节将以金刚石为例子,介绍ARES实空间基组的电子结构自洽计算,并比较PBE与HSE06计算结果。
电子结构自洽计算输入文件
输入文件包含参数文件 ares.in,结构文件 ares.cell 和赝势文件(模守恒赝势,格式为.UPF)。
ares.in 文件内容如下,两个输入文件分别对应PBE泛函和HSE06泛函计算:
INPUT_PARAMETERS
# control
LBAND = T
# system
IXCDF = 0 # PBE
NSMEAR = 1
WSMEAR = 0.01
NADST = 156
# electron
NMITER = 100
ETOL = 1e-6
# mesh
MESH_SPACING = 0.2
KMESH = 3 3 3
KSHIFT = 0 0 0
# pseudopotential
PSEUDO_DIR = ./C.upf
INPUT_PARAMETERS
# control
LBAND = T
# system
IXCDF = 8 # HSE
EXX_ACC = 1
EXX_RANGE_FOCK = 0.106
EXX_RANGE_PBE = 0.106
EXX_ACE_VALENCE_STATES = 28
NSMEAR = 1
WSMEAR = 0.01
NADST = 156
# electron
NMITER = 100
ETOL = 1e-6
# mesh
MESH_SPACING = 0.2
KMESH = 3 3 3
KSHIFT = 0 0 0
# pseudopotential
PSEUDO_DIR = ./C.upf
ares.in 中的参数大致可以分为五个部分
第一部分指定输出控制
LBAND:设置是否输出能量本征值,这里选择输出(TURE)。
第二部分指定系统参数,涉及泛函选择、能带数量、展宽等
IXCDF:设置使用的泛函,这里选择PBE(0)和HSE06(8)。
NSMEAR:设置在布里渊区积分中,处理能带占据数的展宽函数,这里选择高斯展宽(1)。
WSMEAR:设置 smearing 宽度,这里选择 0.01 Ry。
NADST:设置计算的总态数,应大于体系中电子数的一半(本案例体系含256个电子)。
EXX_ACC:设置杂化泛函是否使用自适应压缩算符(ACE)方法加速,这里选择加速(1)。
EXX_RANGE_FOCK:设置Fock精确交换项的范围分离参数,这里选择 0.106 \(\text{bohr}^{-1}\)。
EXX_RANGE_PBE:设置PBE泛函交换项的范围分离参数,这里选择0.106 \(\text{bohr}^{-1}\)。
EXX_ACE_VALENCE_STATES:设置杂化泛函ACE方法计算的空态数,这里选择28(NADST - 体系中电子数的一半)。
第三部分指定电子自洽迭代相关控制参数,迭代步数、收敛参数等
NMITER:设置电子自洽迭代最大次数,这里选择100次。
ETOL:设置电子自洽迭代能量收敛阈值,这里选择1e-6 eV/atom。
第四部分指定计算网格,包括实空间网格密度和倒空间网格
MESH_SPACING:设置实空间网格间距,这里选择0.2 Å(此参数通过采样定理与倒空间能量截断相关,减小网格间距可以系统性提高计算精度)。
KMESH:设置倒空间网格,这里沿三个倒格子基矢方向,每个方向上有3个网格点。
KSHIFT:设置倒空间网格偏移,这里选择0 0 0。
第五部分指定赝势文件
PSEUDO_DIR:设置赝势文件路径,这里选择当前目录下的C.upf文件(./C.upf)。
ares.cell 文件定义了一个由金刚石结构C晶胞构建的2x2x2超胞,共包含64个原子。文件内容示例如下,为清晰起见,仅列出部分原子坐标:
C_64_supercell
1.0
7.1418508899 0.0000000000 0.0000000000
0.0000000000 7.1418508899 0.0000000000
0.0000000000 0.0000000000 7.1418508899
C
64
Direct
0.2500000000 0.0000000000 0.0000000000
0.2500000000 0.0000000000 0.5000000000
0.2500000000 0.5000000000 0.0000000000
0.2500000000 0.5000000000 0.5000000000
0.7500000000 0.0000000000 0.5000000000
... ( remaining 59 atomic coordinates ) ...
结构文件格式与之前的示例相同,包含晶胞参数、原子数和原子坐标。
计算结果分析
根据上述的输入文件,计算完成之后将会得到 ares.result 、 ares.eigen 、 ares.locat 三个文件。
ares.result 为计算的输出文件,包括计算任务信息、输入参数、初始化信息、具体的电子自洽迭代、离子弛豫迭代信息、原子受力、晶格应力、能量等。
ares.eigen 为指定输出的能量本征值文件。
ares.locat 为原子受力文件。
利用能量本征值文件 ares.eigen 可以分析能态密度,本案例中计算了PBE泛函和HSE06泛函的能态密度,结果如下:
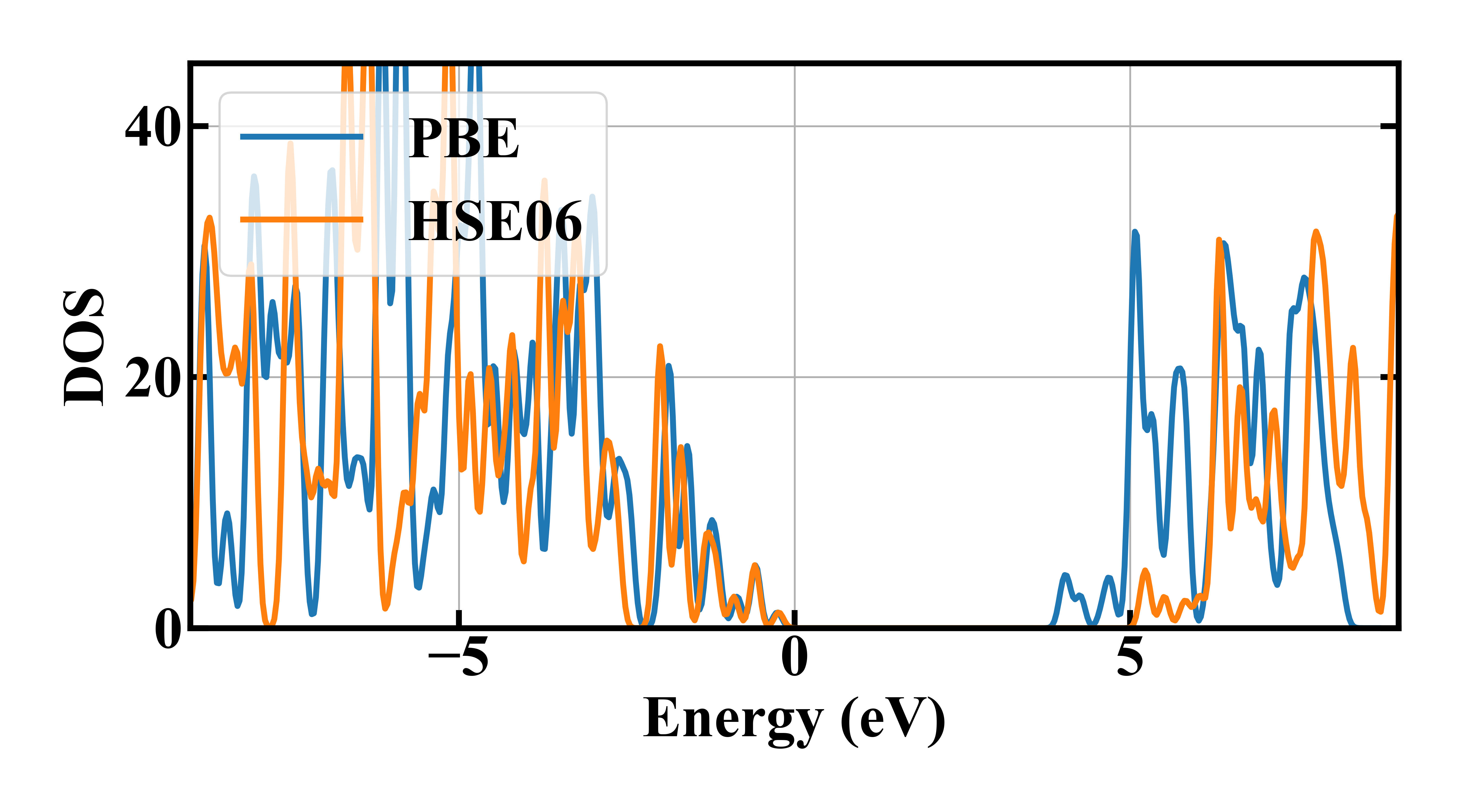
从图中可以看出,PBE泛函计算存在低估带隙的问题,而HSE06泛函计算极大地修正了带隙,与实验值(5.2 eV)更相符。
此外,ARES实空间基组并行扩展性较强,适合计算原子数较多的体系,下面展示了1000个原子的金刚石的能态密度,并且与QE、VASP计算结果进行了对比。
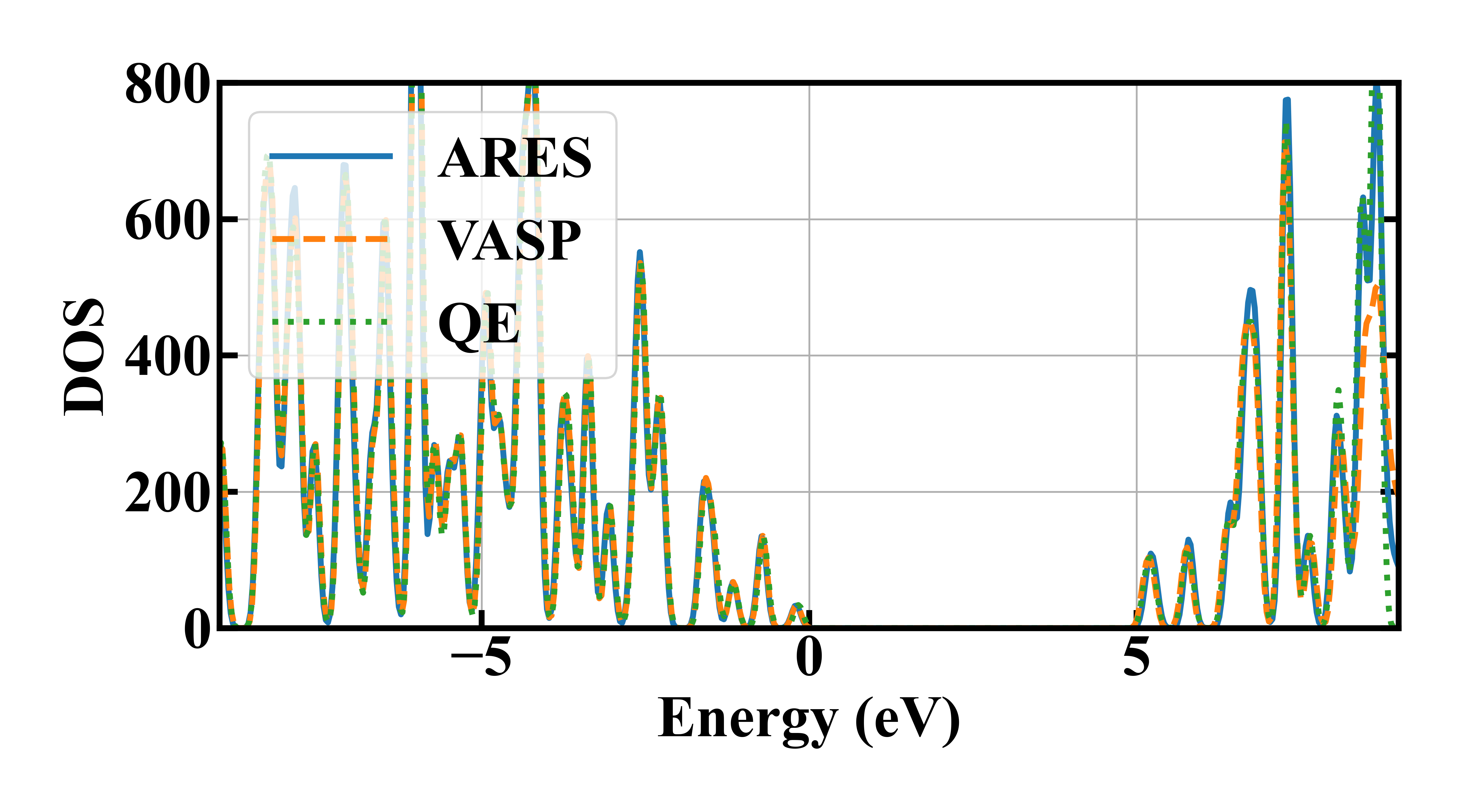
从图中可以看出,ARES计算的能态密度与QE、VASP计算结果高度一致,精度得到验证。
AIMD分子动力学模拟
本节将以单质铝体系为例子,介绍ARES中如何进行分子动力学模拟计算。
AIMD分子动力学模拟输入文件
输入文件包含参数文件 ares.in,结构文件 ares.cell,k点文件 ares.kpoints 和赝势文件(这里是Al.PD04.PBE.UPF)。
结构文件、k点文件和赝势文件的内容与前面介绍的结构弛豫计算相同,这里不再赘述。
ares.cell 文件内容如下:
Al4
1.0
8.0778589249 0.0000000000 0.0000000000
0.0000000000 8.0778589249 0.0000000000
0.0000000000 0.0000000000 8.0778589249
Al
32
Direct
0.000000000 0.009014286 0.004639879
0.001973170 0.000000000 0.493119890
0.000000000 0.507323523 0.002022300
0.004161452 0.490411690 0.509398197
0.506648853 0.000000000 0.000000000
0.493668090 0.000000000 0.500495129
0.498638900 0.495824583 0.002237058
0.492789877 0.495842893 0.497327237
0.000000000 0.255703519 0.243993476
0.000284689 0.251848291 0.740929008
0.002150897 0.743410482 0.241301032
0.008977711 0.759312641 0.756167947
0.496092275 0.241953442 0.253684661
0.498803050 0.242440765 0.749903538
0.490687770 0.758186408 0.245175600
0.503250446 0.746234222 0.750401360
0.250934206 0.000000000 0.259391693
0.255502656 0.008789979 0.757896547
0.251958000 0.508437485 0.241769850
0.243919657 0.490904546 0.746506607
0.747773546 0.000000000 0.256574750
0.747135067 0.000000000 0.750853922
0.742818484 0.506043940 0.241491013
0.759737739 0.505444895 0.743974314
0.240110442 0.256309229 0.004137147
0.254580143 0.255425407 0.491480893
0.247169315 0.742317381 0.007262069
0.252465963 0.746617960 0.491271167
0.746219646 0.246503666 0.004592124
0.752751149 0.257744255 0.499444299
0.742391885 0.754264896 0.005215701
0.751225544 0.755419344 0.499875912
ares.in 文件内容如下:
首先提供一个NVE系综的例子:
# scf
kpar = 4
force_thr = 0.01
diago = dav
sym = 2
iscf_iter = 100
ecutwfc = 816.34
basis = pw
smearing = mp
sigma = 0.01
mixing_type = broyden
mixing_beta = 0.7
scf_thr = 1e-8
scf_energy_thr = 1e-6
pseudo_dir = ./
pseudo_file = Al.PD04.PBE.UPF
# md
cal_type = md
md_method = NVE
ion_temp = 300
timestep = 1.0
nstep = 1000
ares.in 输入参数介绍:
在分子动力学模拟中尽量保留scf自洽相关的计算参数,同时可以适当放宽k-points和ecutwfc等参数以提高计算效率。在此基础上,额外设置md相关参数即可:
cal_type:计算类型,设置为 md 表示分子动力学模拟计算;md_method:分子动力学模拟方法,当前支持NVE、NVT_NH、NVT_NHC和NPT_NHC方法;ion_temp:初始温度,单位为K;timestep:时间步长,单位为fs;nstep:分子动力学模拟总步数;
然后是NVT系综的例子:
# scf
kpar = 4
force_thr = 0.01
diago = dav
sym = 2
iscf_iter = 100
ecutwfc = 816.34
basis = pw
smearing = mp
sigma = 0.01
mixing_type = broyden
mixing_beta = 0.7
scf_thr = 1e-8
scf_energy_thr = 1e-6
pseudo_dir = ./
pseudo_file = Al.PD04.PBE.UPF
# md
cal_type = md
md_method = NVT_NHC
ion_temp = 300
t_init = 300
t_final = 300
timestep = 1.0
nstep = 1000
其中区别在于修改md_method参数,并添加对于控温器的参数。
t_init:控温器初始温度,单位为K;t_final:控温器最终温度,单位为K;
程序运行
准备好所有文件后,在终端中运行以下命令:
mpirun -n np ares > output.out
结果分析
计算完成后,会有采集信息文件 ares.md ,初始化参数文件 ares.result 和轨迹文件 md.traj 生成。
ares.md 记录了体系在分子动力学模拟过程中的能量、温度、压强等信息,可以用来分析体系的热力学性质。其中在文件开头列出了各个参数的含义及其单位,在后续结果输出中以每一个离子步为一单元块进行输出。
具体开头如下:
Description:
Desc_TIO: Ionic temperature. Unit=Kelvin
Desc_TEN: Total energy. TEN = KEN + FEN. Unit=Ha/atom
Desc_KEN: Ionic kinetic energy. Unit=Ha/atom
Desc_KENIG: Kinetic energy: 3/2 N k T of ideal gas at temperature T = TIO. Unit=Ha/atom
where N = number of particles, k = Boltzmann constant
Desc_FEN: Free energy F = U - TS. FEN = UEN + TSEN. Unit=Ha/atom
Desc_UEN: Internal energy. Unit=Ha/atom
Desc_TSEN: Electronic entropic contribution -TS to free energy F = U - TS. Unit=Ha/atom
Desc_STRESS: Stress, excluding ion-kinetic contribution. Unit=GPa*Bohr**3
Desc_STRESS_TOT: Stress, inluding ion-kinetic contribution. Unit=GPa*Bohr**3
Desc_PRES: Pressure, struct pressure. Unit=GPa
Desc_PRESTO: Pressure, excluding kinetic contribution. Unit=GPa
Desc_PRESIG: Pressure N k T/V of ideal gas at temperature T = TIO. Unit=GPa
where N = number of particles, k = Boltzmann constant, V = volume
Desc_AVGV: Average of the speed of all ions of the same type. Unit=Bohr/atu
Desc_MAXV: Maximum of the speed of all ions of the same type. Unit=Bohr/atu
Desc_MIND: Minimum of the distance of all ions of the same type. Unit=Bohr
提取文件中的数据,可以分析体系的热力学性质,如温度、能量、压强等随时间的变化情况。
例如在NVE模拟中,我们比较关注能量的波动,通过提取 ares.md 文件中的总能量(TEN)数据,可以绘制能量随时间的变化曲线:
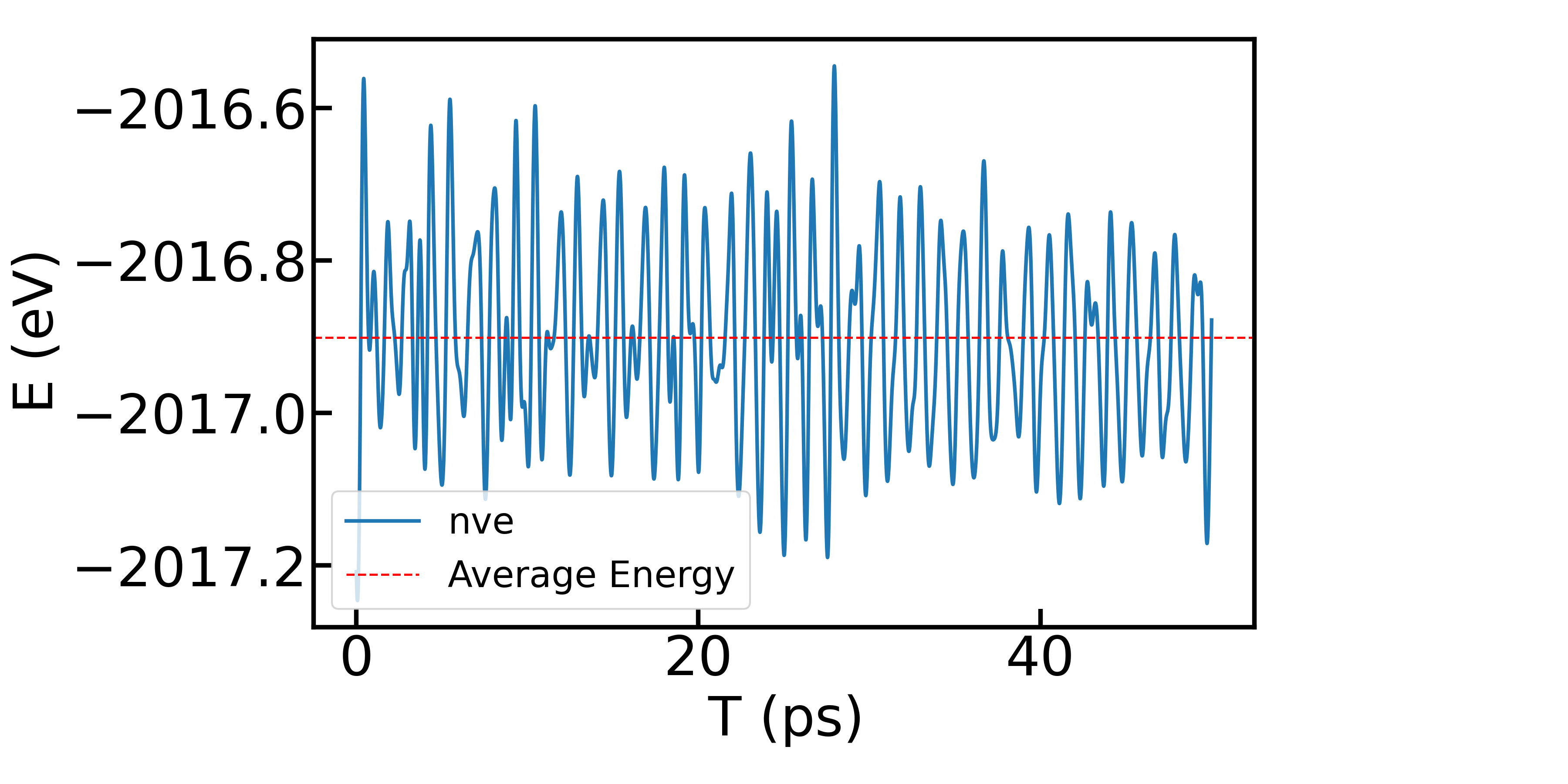
在NVT模拟中,我们比较关注温度的波动,通过提取 ares.md 文件中的离子温度(TIO)数据,可以绘制温度随时间的变化曲线:
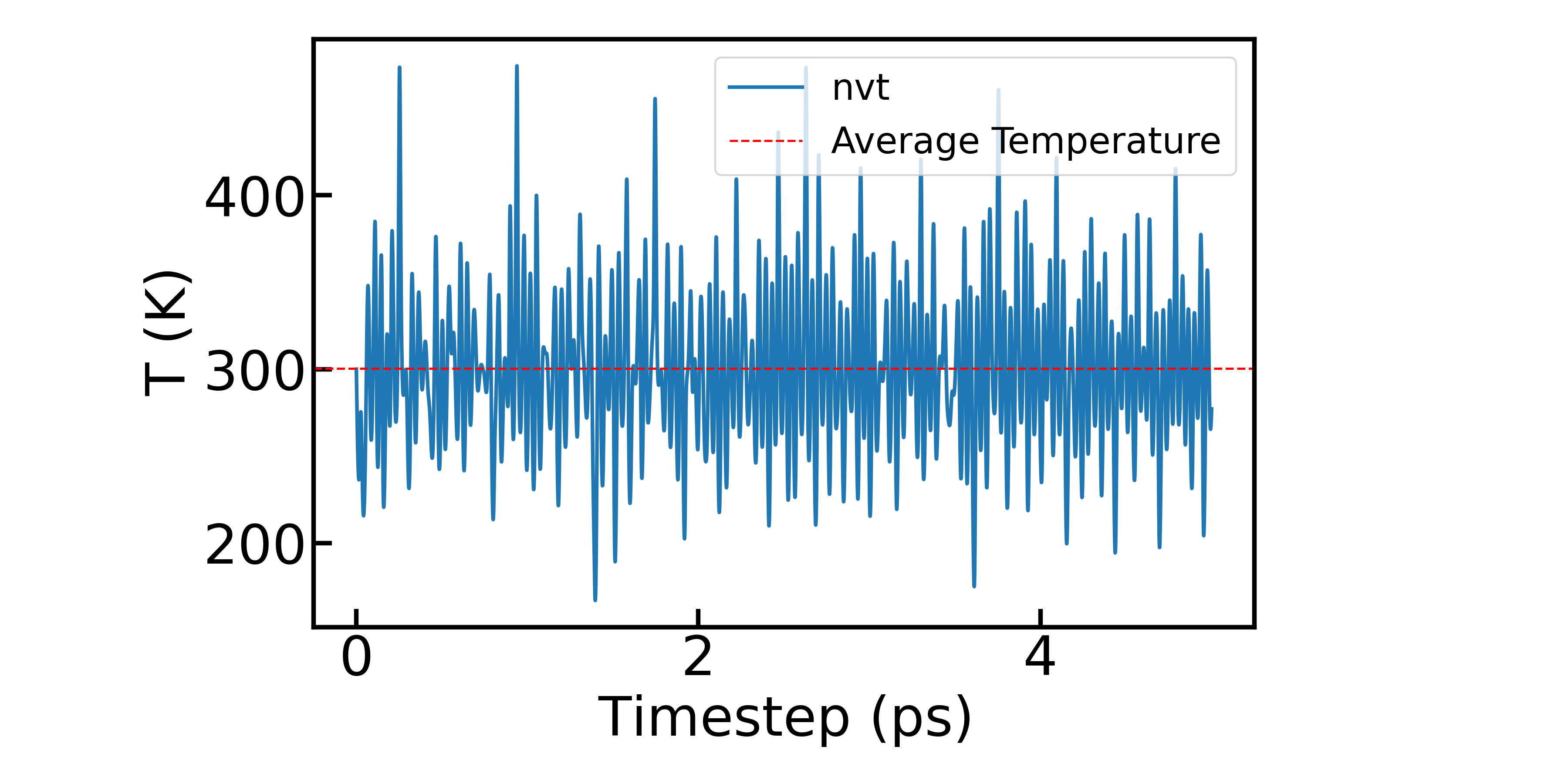
ares.result 记录了体系的初始结构信息和从 ares.in 文件中读取的输入信息,包括原子种类、原子数目、晶胞参数、输入设置等。
md.traj 记录了体系在分子动力学模拟过程中的原子坐标信息,可以用来分析体系的结构演化过程。轨迹文件可以用OVITO等可视化软件进行可视化。
MLMD分子动力学模拟
本节以AlScN体系为例子,介绍ARES中如何进行基于机器学习势的分子动力学模拟计算。
MLMD分子动力学模拟输入文件
输入文件包含参数文件 ares.in,结构文件 ares.cell 和机器学习势文件(这里是model-test)。
结构文件同前面介绍的结构弛豫计算相同,本例子中采用了包含15360原子数的 \(\mathrm{Al_{0.75}Sc_{0.25}N}\) 结构,机器学习势文件参考ACNN软件生成。
ares.in 文件内容如下:
PRINT_FORCES 0
PRINT_ATOMS 1
PRESS 1
LSTRESS 1
UNIT 1
SUPERCELL 1 1 1
# MD
MD_FLAG 1
MD_METHOD NVE
MD_TIMESTEP 1
ION_TEMP 300
# potential
EF_METHOD ML ./model-test
#output
PRINT_TRAJ_FQ 10
PRINT_MDOUT_FQ 10
MD_NSTEP 10000
ares.in 输入参数介绍:
PRINT_FORCES:是否输出力,0:不输出,1:输出;PRINT_ATOMS:是否输出原子坐标,0:不输出,1:输出;PRESS:是否计算压强,0:不计算,1:计算;LSTRESS:是否计算应力张量,0:不计算,1:计算;UNIT:单位选择,0:原子单位,1:国际单位;SUPERCELL:超胞倍数,三个整数分别对应a、b、c方向的倍数,根据 ares.cell 的结构在软件内直接生成超胞进行模拟;MD_FLAG:是否进行分子动力学模拟,0:不进行,1:进行;MD_METHOD:分子动力学模拟方法,当前支持NVE、NVT_NH、NVT_NHC、NVT_MNHC、NVT_L、NVT_BDP、NVT_VV、NPT_NHC、PIMD_NVE、PIMD_NVT_NHC、PIMD_NVT_MNHC、PIMD_NPT_NHC、PIMD_NPT_MNHC方法;MD_TIMESTEP:时间步长,单位为fs;ION_TEMP:初始温度,单位为K;EF_METHOD:势函数类型及文件位置,接机器学习势文件相对路径或者绝对路径;PRINT_TRAJ_FQ:轨迹文件输出频率,单位为步;PRINT_MDOUT_FQ:ares.md 文件输出频率,单位为步;MD_NSTEP:分子动力学模拟总步数;
如果需要进行NVT系综的模拟,则需要修改``MD_METHOD``参数,并添加对于控温器的参数。
MD_METHOD NVT_MNHC
ION_TEMP_END 300
PTHERMO 10
NNOS 4
ION_TEMP_END:控温器的最终温度,单位为K;PTHERMO:温度调节频率,单位和时间步长相同;NNOS:Nose-Hoover链的长度;
如果需要进行NPT系综的模拟,则需要修改``MD_METHOD``参数,并添加对于控温器和控压器的参数。
NPT_TRIC 1
TARGET_PRESSURE 0.0001 0.0001
NPT_TRIC:是否考虑晶胞形变,0:不考虑,1:考虑;TARGET_PRESSURE:三个方向的目标压强,单位为GPa;(如果需要对三个方向分别设置,则使用X_PRESSURE,Y_PRESSURE,Z_PRESSURE)
程序运行
准备好所有文件后,在终端中运行以下命令:
mpirun -n np ares > output.out
结果分析
计算完成后,会有采集信息文件 ares.md ,初始化参数文件 ares.result 和轨迹文件 md.traj 生成。
首先展示NVE系综下体系能量的变化:
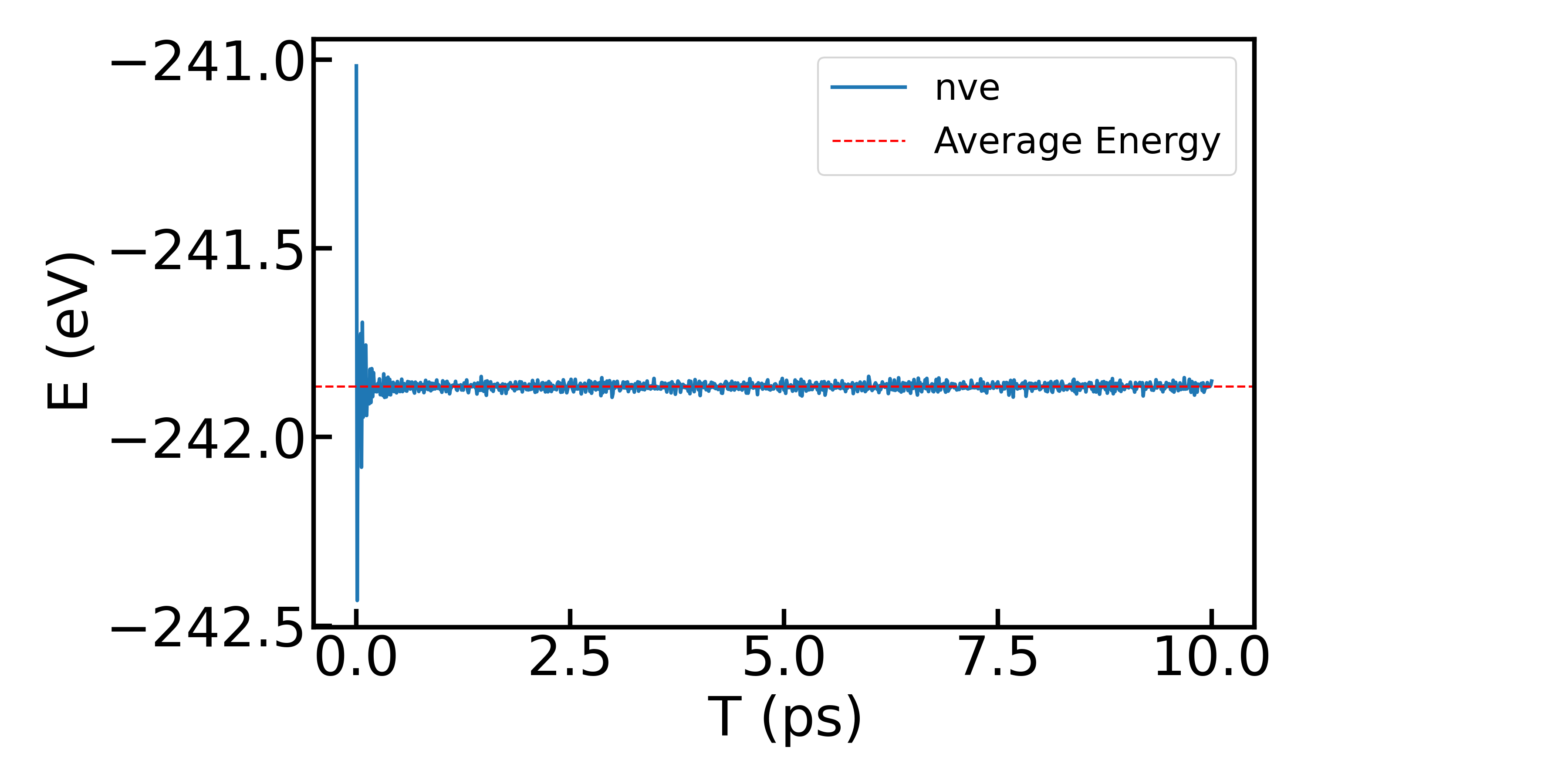
然后展示NVT系综下体系温度的变化:
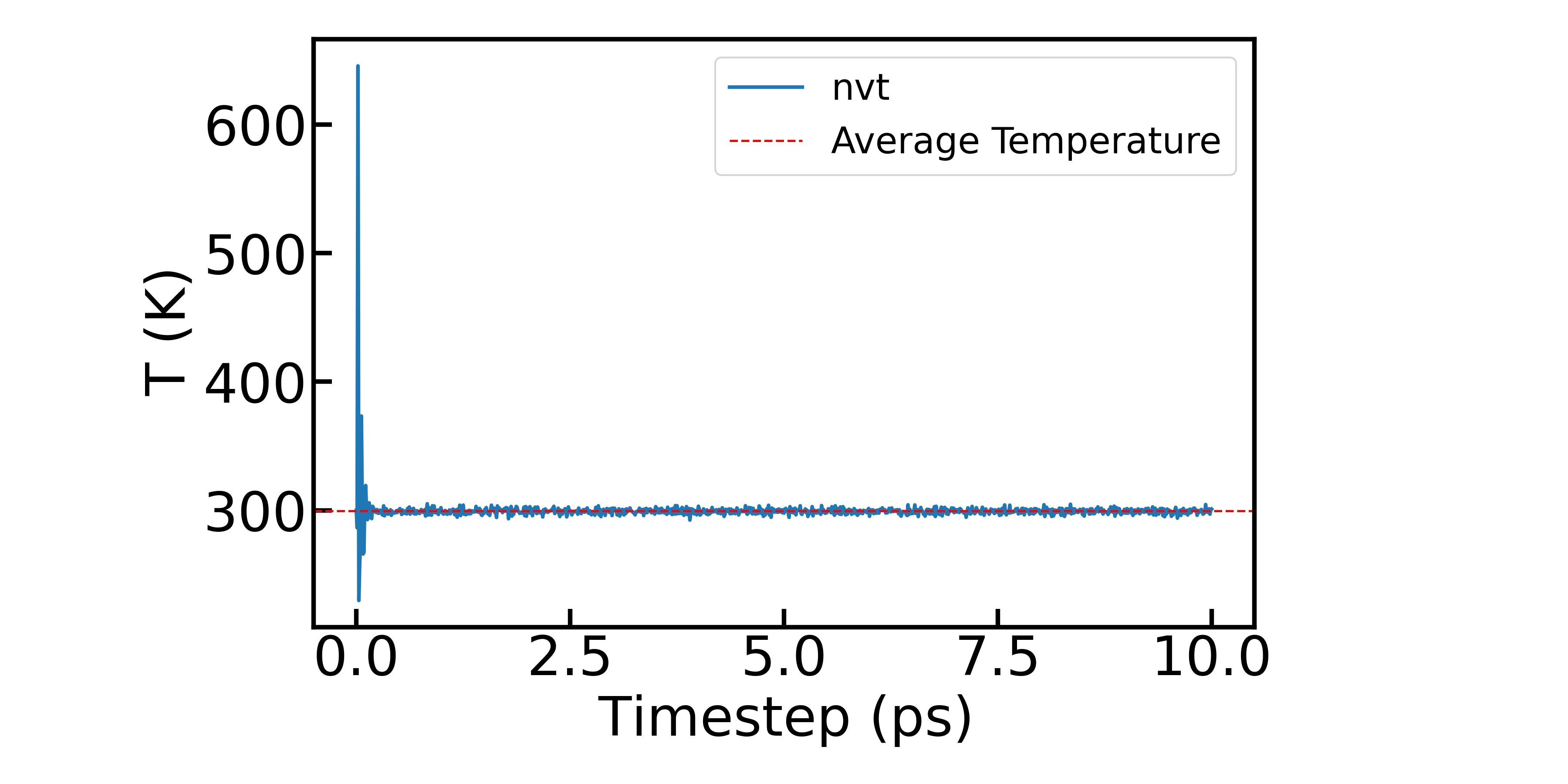
最后展示NPT系综下,体系的能量、温度、压强随时间的变化情况:
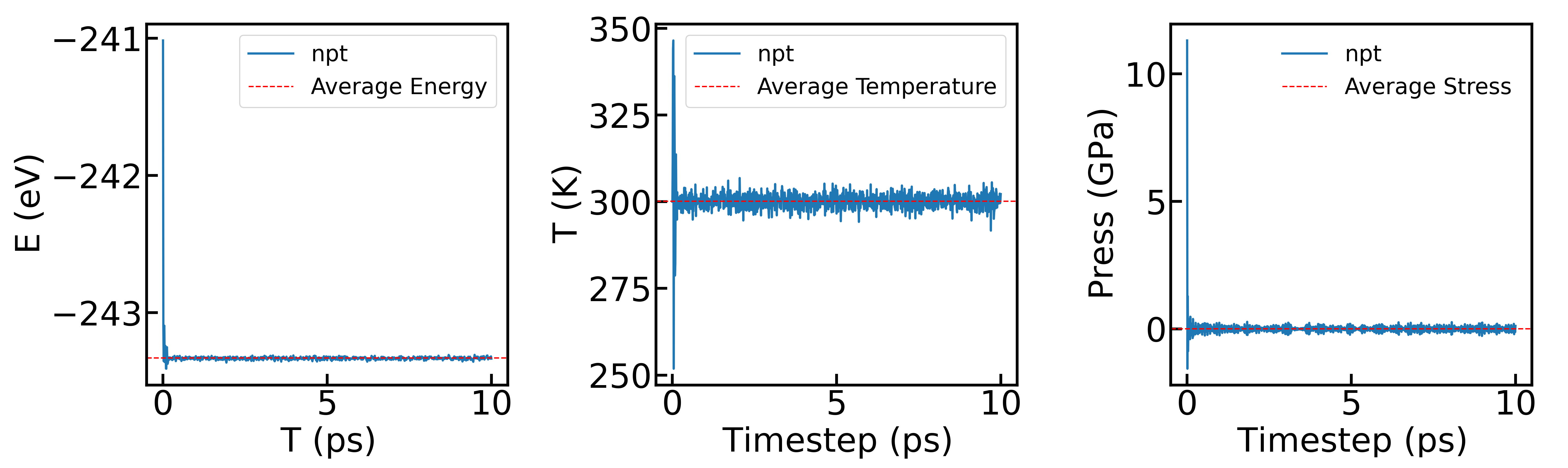
可以明显看到,随着模拟尺度的增大,体系的温度和压强波动幅度明显减小,能量也趋于更加平稳。
格式和表示信息都和AIMD模块一致,这里就不做过多赘述。
晶格动力学计算
晶格动力学(lattice dynamics)是研究晶体中原子振动及其相互作用的理论框架。在晶体中,原子并非静止地排列在平衡位置上,而是在热扰动或外部激发下不断地振动。这些集体的、相干的原子振动可量子化为准粒子——声子(phonon)。
声子是晶体振动的量子,是晶格热学、力学和输运性质的基本载体。通过研究声子,可以深入理解和预测材料的热导率、比热、热膨胀、相变行为、电子-声子耦合效应等关键物理性质。
有限位移法(finite displacement method)是计算声子最常用、最直观的数值方法之一。它通过在平衡构型附近对原子施加微小扰动,并计算由此产生的力响应,从而获得力常数矩阵(force constant matrix):
其中, \(F_i^\alpha\) 是第 \(i\) 个原子在 \(\alpha\) 方向上的力, \(u_j^\beta\) 是第 \(j\) 个原子在 \(\beta\) 方向上的位移。通过对所有原子及其相应的位移方向进行系统地扰动和力计算,可以构建完整的力常数矩阵。
之后,通过对力常数矩阵进行傅里叶变换,得到动量空间中的动力学矩阵(dynamical matrix):
其中, \(m_i\) 和 \(m_j\) 分别是第 \(i\) 和第 \(j\) 个原子的质量, \(\mathbf{R}\) 是晶格矢量, \(\mathbf{q}\) 是波矢。对角化动力学矩阵,可以得到声子的频率和本征向量,进一步求得声子色散关系与相关热力学量。
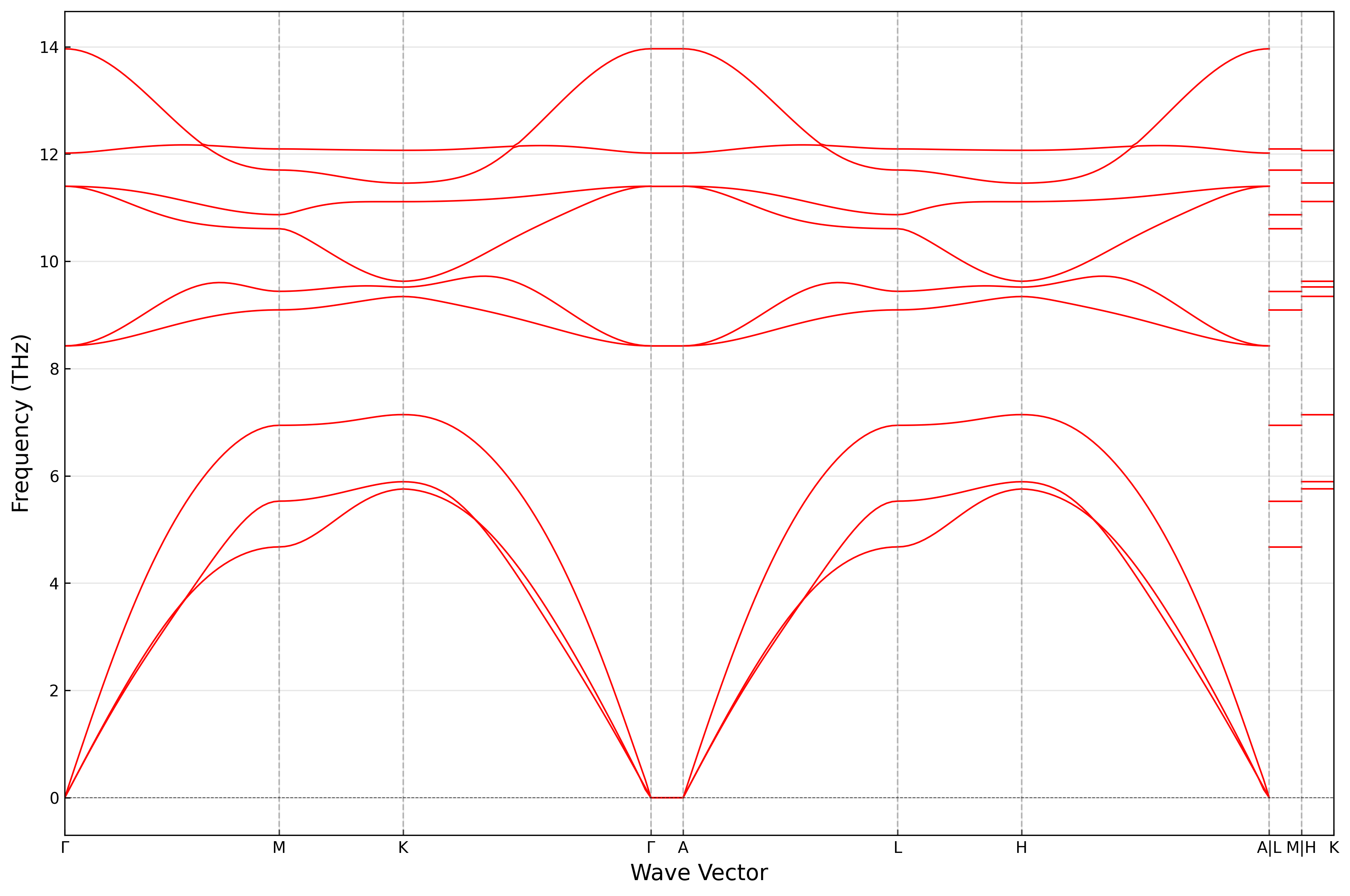
在 ARES-EPC 中,可以使用两种不同的方法来计算声子: 对角超胞法 和 非对角超胞法。对于对角超胞法,其主要侧重于力常数矩阵是否收敛,即在超胞边界处力常数是否衰减至零,但并未考虑特定 \(\mathbf{q}\) 点的周期性边界条件: \(\mathbf{q}\cdot\mathbf{R} = n\) 。 而非对角超胞法则是针对特定的 \(\mathbf{q}\) 点,构建满足该 \(\mathbf{q}\) 点周期性边界条件的最小超胞,从而更高效地计算力常数矩阵,但未必保证力常数矩阵的收敛性。两者各有优缺点,在计算精度上,通常对角超胞法更为可靠,而在计算效率上,非对角超胞法则更具优势。 具体理论细节可参考文献:https://arxiv.org/abs/2503.11013。
软件的工作流程如下图所示:
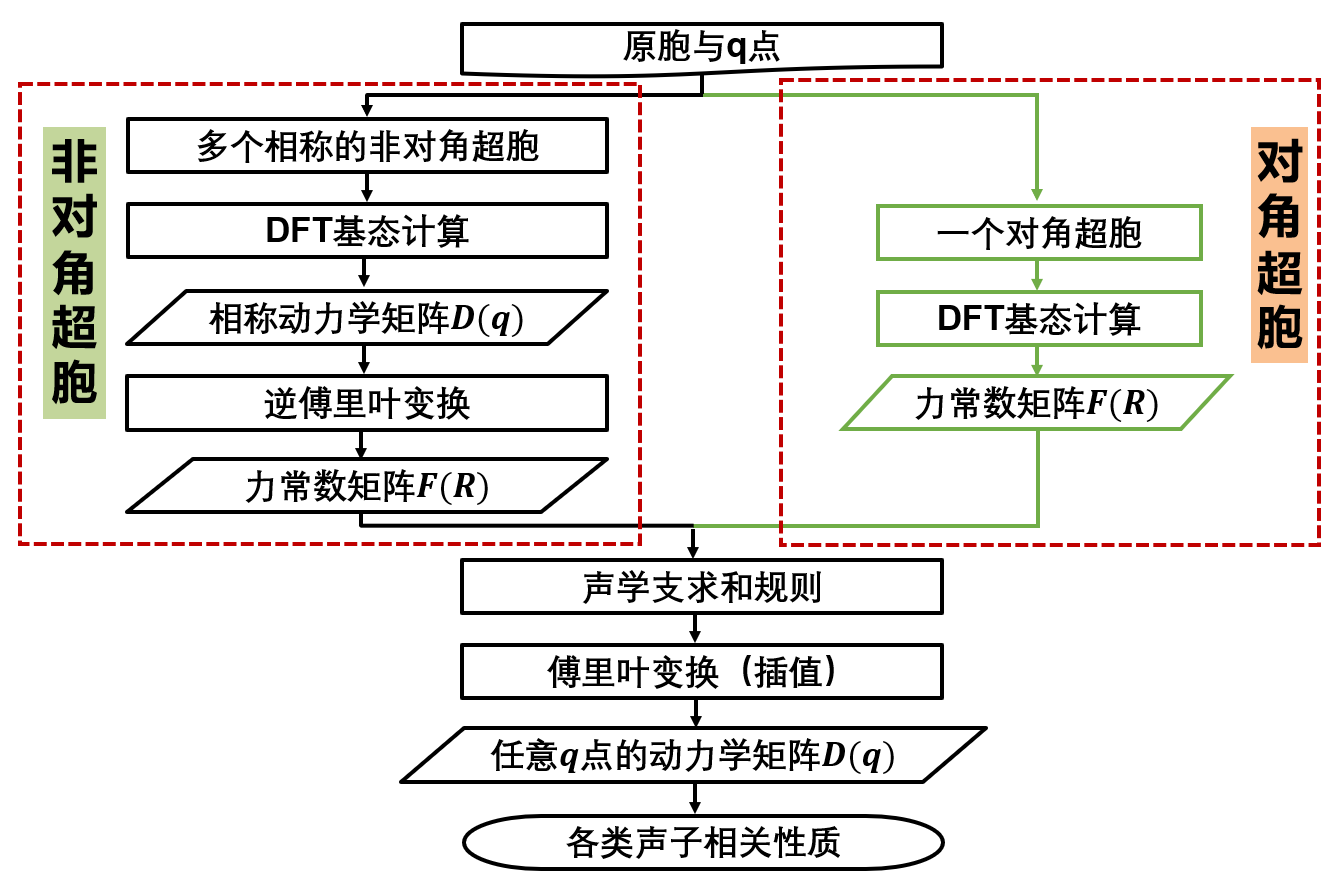
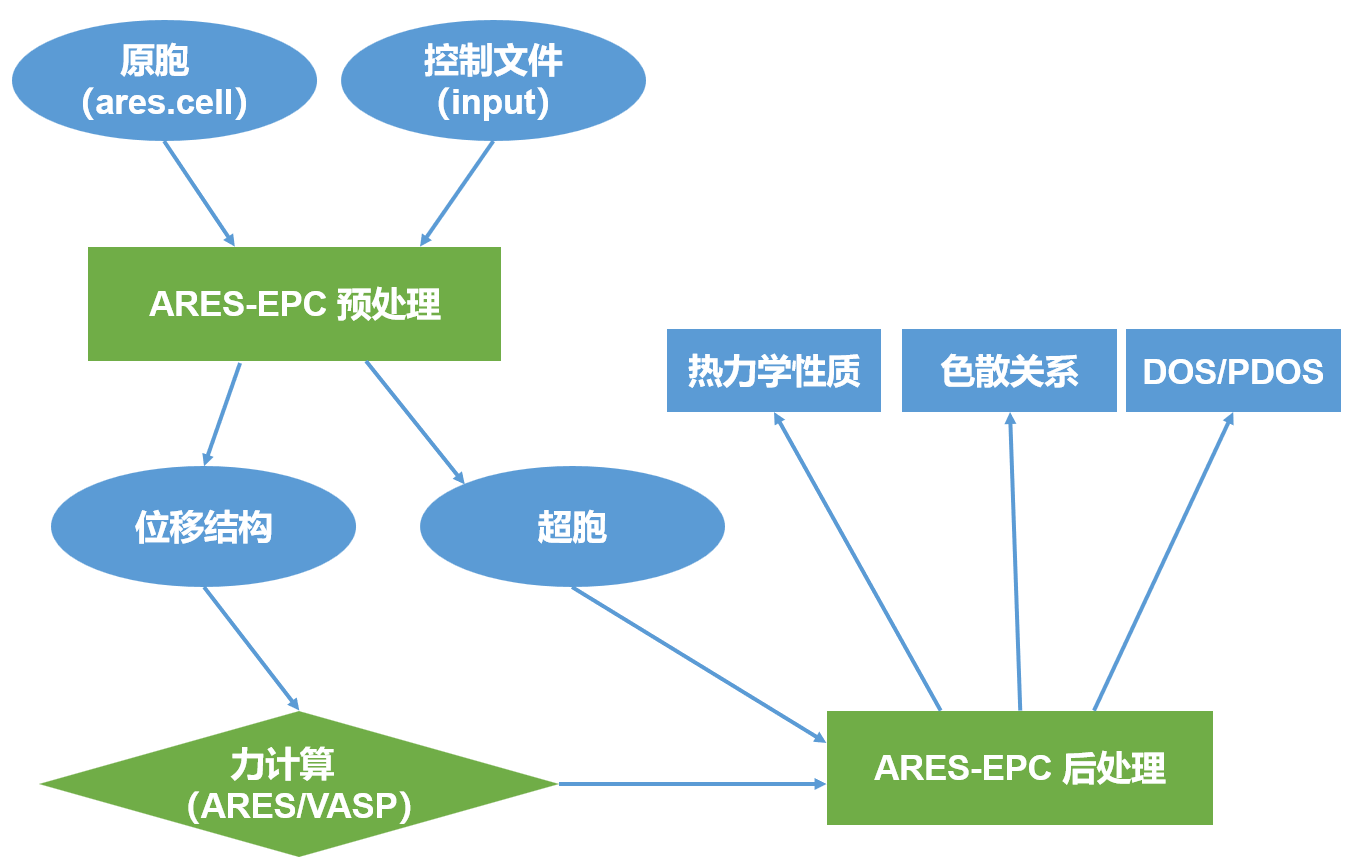
下面将分别以金刚石与层状 \(\mathrm{MoS_2}\) 为例,介绍这两种方法的具体使用。
1. 对角超胞法
示例输入文件input内容如下:
calcType = "phonon"
calcMethod = "diagonal"
calcMode = "separate"
calcRun = "preprocess"
structFile = "ares.cell"
findPrimitiveCell = true
idealizeStructure = true
dftCode = "ares"
dftForceFile = "ares_output.dat"
superMatrix = [4 0 0
0 4 0
0 0 4]
useCentralDiff = true
useSymmetry = true
useTimeReversalSymmetry = true
symTolerance = 1e-5
dispAmplitude = 0.01
verbosity = "high"
参数说明:
参数名 |
类型 |
默认值 |
说明 |
|---|---|---|---|
|
|
|
计算类型。
|
|
|
|
计算方法。
|
|
|
|
计算模式。
|
|
|
|
当前运行阶段。
|
|
|
|
输入结构文件。 使用 ARES 格式的 |
|
|
|
是否自动寻找并使用原胞(primitive cell)。 |
|
|
|
是否将结构理想化(将原子移动至对称位置)。 |
|
|
|
所使用的第一性原理计算软件。 目前支持 |
|
|
|
第一性原理计算所得的力文件。 对于 ARES,使用 对于 VASP,使用 |
|
|
|
超胞矩阵,用于定义沿各晶轴的超胞扩展倍数。 |
|
|
|
是否使用中心差分法(central difference method)计算力常数。 |
|
|
|
是否利用空间群对称性简化计算。 |
|
|
|
是否利用时间反演对称性简化计算。 |
|
|
|
对称性判断容差(用于识别等价原子与对称操作)。 |
|
|
|
原子位移幅度(单位:Å),例如 |
|
|
|
输出信息的详细程度,可选 |
运行程序生成超胞位移结构:
ARES-EPC/main -in input -out output
生成的文件结构如下:
当前目录/
├── SYMMETRY
├── BZQMESH
├── IBZQMESH
├── PRIMCELL
├── SUPERMATRICES
├── IBZTOSUP
├── output
└── SUPERCELL.0/
├── COMMENSURATE_QPOINTS
├── SPOSCAR
├── SYMMETRY
├── SUPERMATRIX
├── DISPLACEMENT
└── DISP.*/
└── ares.cell
运行第一性原理计算软件(例如 ares)计算各超胞位移结构的力。在计算完所有位移结构受力后,便可以进行后处理,求解声子性质。
修改 input 文件中的参数:
# all input parameters for ares-epc
calcType = "phonon"
calcMethod = "diagonal"
calcMode = "seperate"
calcRun = "postprocess"
structFile = "ares.cell"
findPrimitiveCell = true
idealizeStructure = true
dftCode = "ares"
dftForceFile = "ares_output.dat"
superMatrix = [4 0 0
0 4 0
0 0 4]
useCentralDiff = true
useSymmetry = true
useTimeReversalSymmetry = true
symTolerance = 1e-5
dispAmplitude = 0.01
qMeshFine = [4, 4, 4]
qMeshFineShift = [0, 0, 0]
minPhononFreq = 0.0
acousticSumRule = "simple"
calcPhononSpectrum = true
calcPhononThermal = true
tempRange = [0, 1000, 10]
verbosity = "high"
saveForceConstants = true
saveDynamicMatrix = true
savePhononFreq = true
savePhononEigvec = true
新增参数说明:
参数名 |
类型 |
默认值 |
说明 |
|---|---|---|---|
|
|
|
当前运行阶段。选择 |
|
|
|
用于计算声子谱的细化布里渊区网格密度。 |
|
|
|
布里渊区网格的平移矢量。 |
|
|
|
声子频率的最小阈值(单位:THz),用于过滤掉数值噪声引起的负频率。 |
|
|
|
声子力常数矩阵的修正方法。选择 |
|
|
|
是否计算并输出声子色散关系到文件 |
|
|
|
是否计算并输出声子热力学性质到文件 |
|
|
|
计算声子热力学性质的温度范围与步长,例如 |
参数名 |
类型 |
默认值 |
说明 |
|---|---|---|---|
|
|
|
是否保存力常数矩阵到文件 |
|
|
|
是否保存动态矩阵到文件 |
|
|
|
是否保存声子频率到文件 |
|
|
|
是否保存声子本征向量到文件 |
运行程序进行后处理,计算声子性质:
mpirun -np n ARES-EPC/main -in input -out output
计算完成后,文件夹下将生成如下文件:
当前目录/
├── BZQMESH_FINE
├── IBZQMESH_FINE
├── FORCE_CONSTANTS
├── DYNAMICMATRIX_FINE
├── EIGENVECTORS_FINE
├── PHONONFREQS_FINE
├── PHONON_SPECTRUM
├── PHONON_SPECTRUM.MODE
├── THERMAL_PROPERTIES
└── output
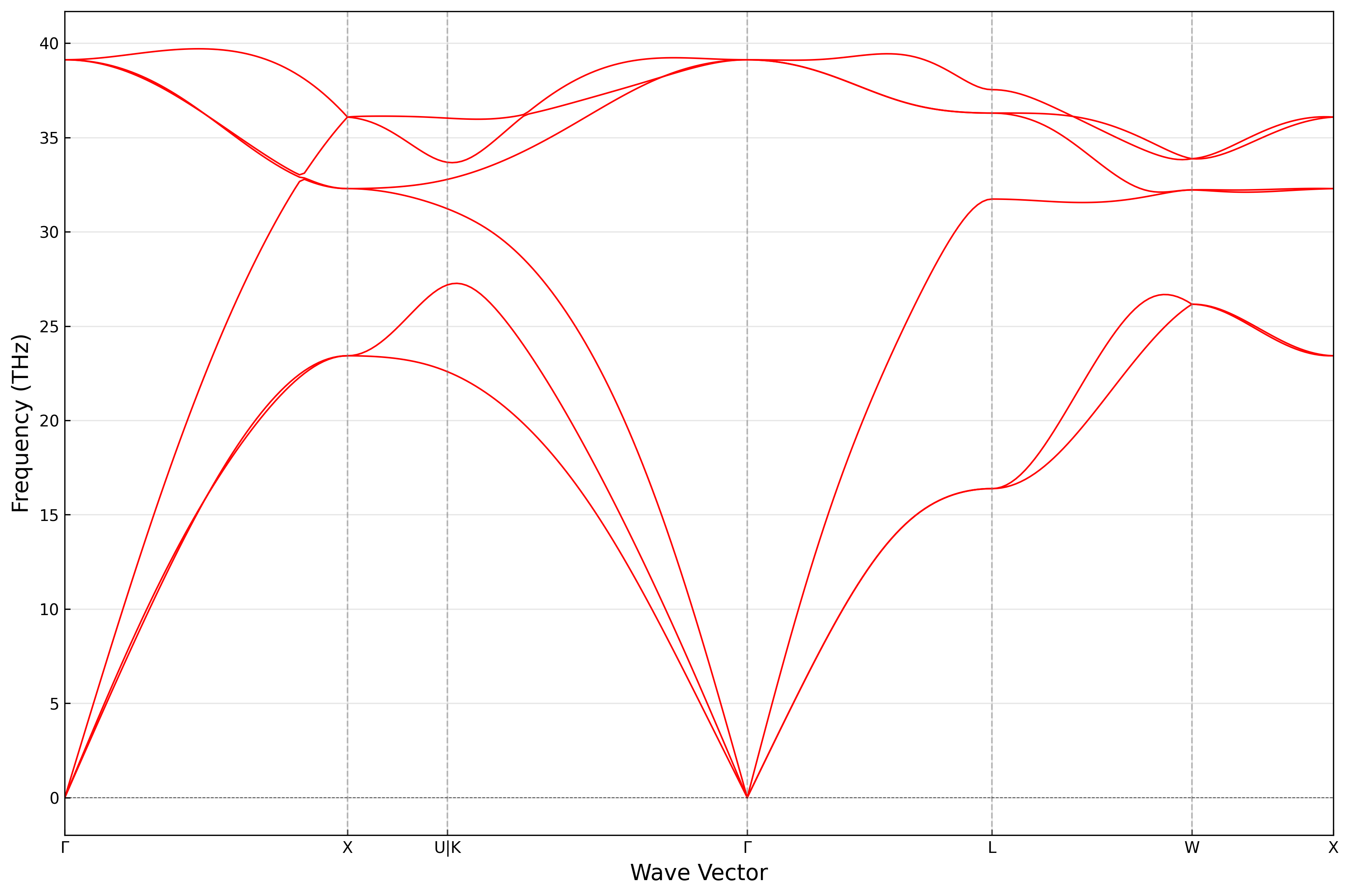
其中,PHONON_SPECTRUM文件包含声子色散关系数据,THERMAL_PROPERTIES文件包含声子热力学性质数据。 调用ARES-EPC/utils/plot_phonon_spectrum.py脚本绘制声子色散关系:
python ARES-EPC/utils/plot_phonon_spectrum.py
结果如图所示:
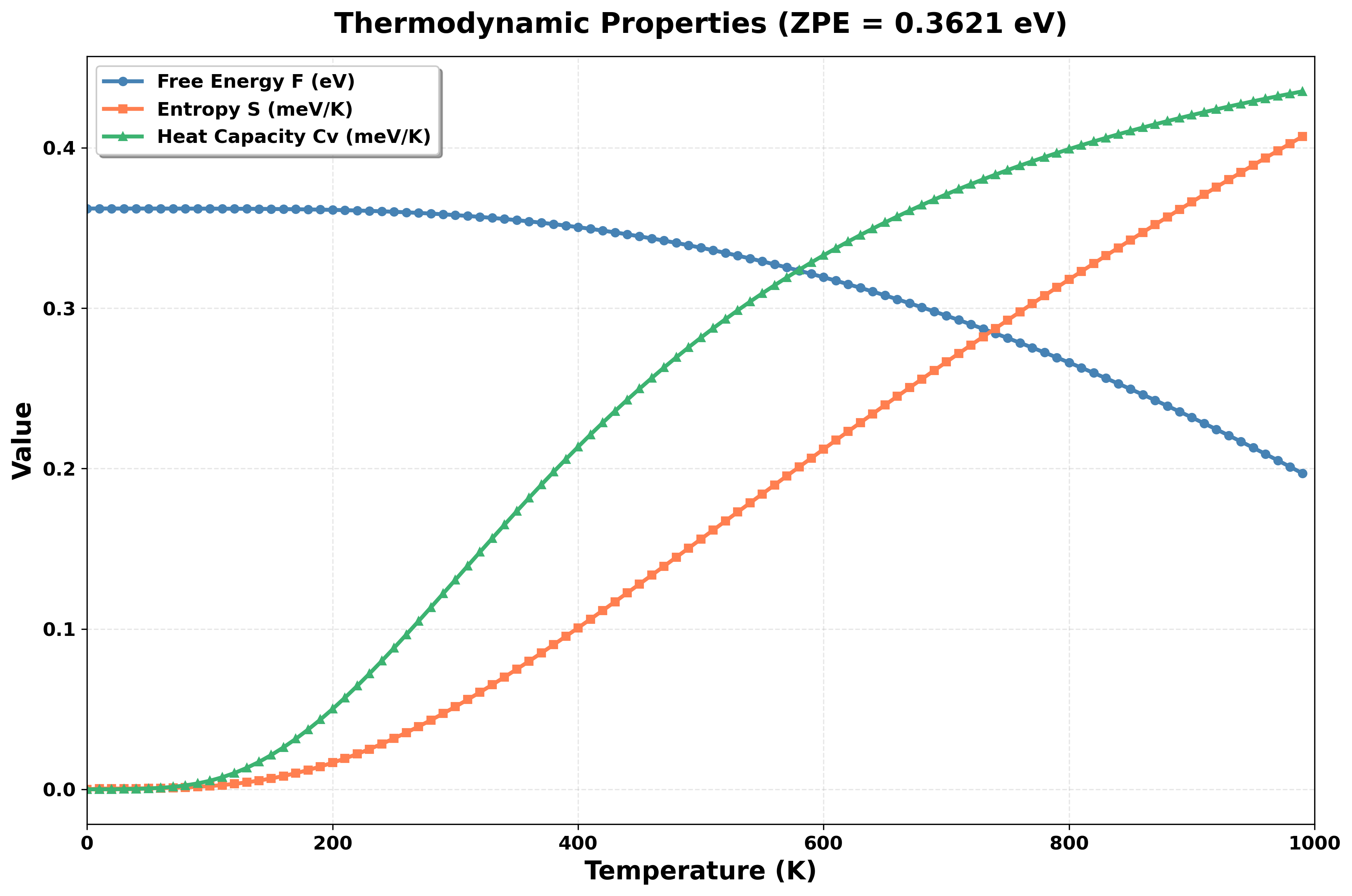
调用ARES-EPC/utils/plot_thermal_properties.py脚本绘制声子热力学性质:
python ARES-EPC/utils/plot_thermal_properties.py
结果如图所示:
2. 非对角超胞法
非对角超胞法输入参数与对角超胞法基本相同,其区别仅在于超胞的构建不由用户指定扩包的倍数,而是由程序根据所需的q点自动生成最小超胞。下面以层状 \(\mathrm{MoS_2}\) 为例,介绍非对角超胞法的具体使用。
示例输入文件input内容如下:
# all input parameters for ares-epc
calcType = "phonon"
calcMethod = "nondiagonal"
calcMode = "seperate"
calcRun = "preprocess"
structFile = "ares.cell"
findPrimitiveCell = true
idealizeStructure = true
dftCode = "ares"
dftForceFile = "ares_output.dat"
useCentralDiff = true
useSymmetry = true
useTimeReversalSymmetry = true
symTolerance = 1e-5
minSupercellLength = 0
qMesh = [4, 4, 1]
dispAmplitude = 0.01
qMeshFine = [10, 10, 10]
minPhononFreq = 0.0
acousticSumRule = "simple"
calcPhononThermal = true
tempRange = [200, 700, 20]
verbosity = "high"
saveForceConstants = true
saveDynamicMatrix = true
savePhononFreq = true
savePhononEigvec = true
新增参数说明:
参数名 |
类型 |
默认值 |
说明 |
|---|---|---|---|
|
|
|
最小超胞边长(单位:Å)。如果生成的非对角超胞边长小于该值,则增加超胞长度以满足要求。 |
|
|
|
用于生成非对角超胞的布里渊区网格密度。超胞将满足改网格点的周期性边界条件。 |
运行程序生成非对角超胞位移结构:
mpirun -np n ARES-EPC/main -in input -out output
生成的文件结构如下:
当前目录/
├── SYMMETRY
├── BZQMESH
├── IBZQMESH
├── PRIMCELL
├── SUPERMATRICES
├── IBZTOSUP
├── output
└── SUPERCELL.*/
├── COMMENSURATE_QPOINTS
├── SPOSCAR
├── SYMMETRY
├── SUPERMATRIX
├── DISPLACEMENT
└── DISP.*/
└── ares.cell
此时将产生多个非对角超胞文件夹(SUPERCELL.*),每个文件夹对应一个不同的非对角超胞结构。接下来的步骤与对角超胞法相同:使用第一性原理计算软件计算各超胞位移结构的力,然后进行后处理求解声子性质。
修改 input 文件中的的calcRun参数,改为 postprocess,然后运行程序进行后处理,计算声子性质:
ARES-EPC/main -in input -out output
python ARES-EPC/utils/plot_phonon_spectrum.py
结果如图所示: